
More Information
Submitted: 03 June 2020 | Approved: 14 July 2020 | Published: 16 July 2020
How to cite this article: Smith GA. The mechanisms of cardiac myopathies, a kinetics approach: Leading review. J Cardiol Cardiovasc Med. 2020; 5: 141-152.
DOI: 10.29328/journal.jccm.1001101
ORCiD: orcid.org/0000-0002-3959-3523
Copyright License: © 2020 Smith GA. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Cardiac myosin binding cardiac protein-C (cMyBP-C); Cardiac troponin-I (cTnI); Cardiac troponin T (cTnT); Cardiac troponin C (cTnC); Calcium dependency; Magnesium ATP (MgATP); Myofilaments; ATPase; Contraction; Ca2+ cooperativity; Hill coefficient (nH); Sarcomere growth and loss

The mechanisms of cardiac myopathies, a kinetics approach: Leading review
Gerry A Smith*
Department of Biochemistry, University of Cambridge, Tennis Court Rd. Cambridge CB2 1QW, UK
*Address for Correspondence: Gerry A Smith, Department of Biochemistry, University of Cambridge, Tennis Court Rd. Cambridge CB2 1QW UK, Tel: 44 1223 515394; Email: gas1000@cam.ac.uk
The normal adult heart is a well maintained machine that has a mechanism for growth replacement of the sarcomere that is lost by natural degeneration. This process ensures the heart has the strength of contraction to function correctly giving blood supply to the whole body. Some of the force of contraction of the sarcomere is transmitted to its major protein titin where its strength results in unfolding of a flexible section and release of a growth stimulant. The origin of all the cardiomyopathies can be traced to errors in this system resulting from mutations in a wide variety of the sarcomeric proteins. Too much or chronic tension transfer to titin giving increased growth resulting in hypertrophic cardiomyopathy (HCM) and too little leading to muscle wastage, dilated cardiomyopathy (DCM). HCM can ultimately lead to sudden cardiac death and DCM to heart failure. In this paper I show (1) a collection of the tension/ATPase calcium dependencies of cardiac myofibrils that define the mechanism of Ca2+ cooperativity. (2) I then reintroduce the stress/strain relationship to cardiomyopathies. (3) I then review the cardiomyopathy literature that contains similar Ca2+ dependency data to throw light on the mechanisms involved in generation of the types of myopathies from the mutations involved. In the review of cardiomyopathy there are two sections on mutations, the first dealing with those disrupting the Ca2+ cooperativity, i.e. the Hill coefficient of activation, leading to incomplete relaxation in diastole, chronic tension, and increased growth. Secondly dealing with those where the Ca2+ cooperativity is not affected giving either increased or decreased tension transfer to titin and changes in sarcomere growth.
These considerations have confirmed that the inhibitory function of cardiac troponin-I (cTnI) is part of a concerted process with the regulatory cardiac myosin binding protein-C (cMyBP-C) to block the use of myosin light chain (MLC) bound magnesium adenosine triphosphate (MgATP) from being used as cross-bridge substrate until the magnesium bound is exchanged for calcium. This Ca2+ dependency added to the trigger troponin-C (cTnC) Ca2+ binding is the origin of the Ca2+ cooperativity (Figure 1).
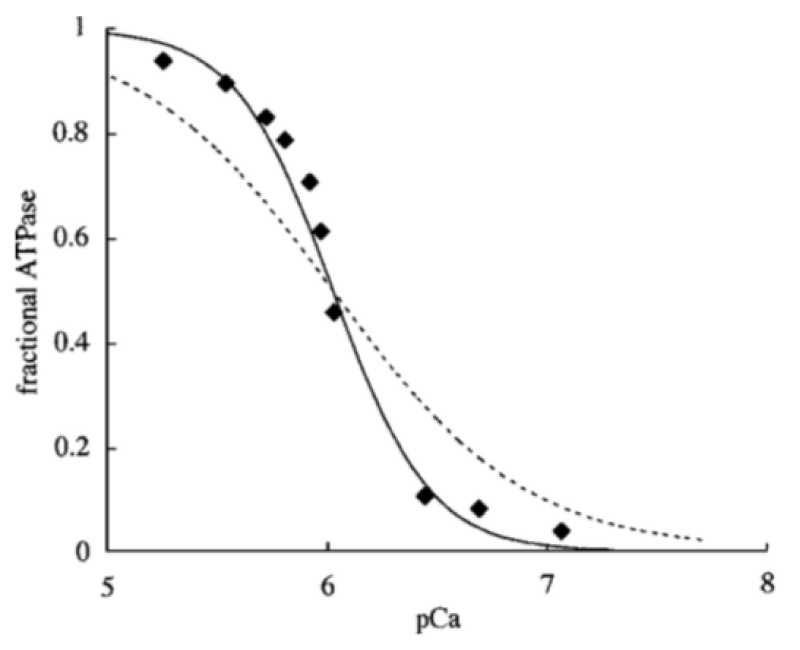
Figure 1: Ca2+ dependence of myofibrillar ATPase. Data points (◆) are reproduced, with permission, from Holroyde, et al. [1]. Lines represent best fits for 1:1 Ca2+ (dotted) and 2:1 Ca2+ (solid) binding schemes. Half maximal is 6.1 M. Smith, et al. [2].
The Ca2+ activation of the myofibrils is cooperative
The conclusion is that two Ca2+ binding sites are occupied cooperatively for activation of the cross bridge ATPase. The ATP usage is unimolecular.
The measurable binding of Ca2+ to the myofibrils is unimolecular, not cooperative (Figure 2).
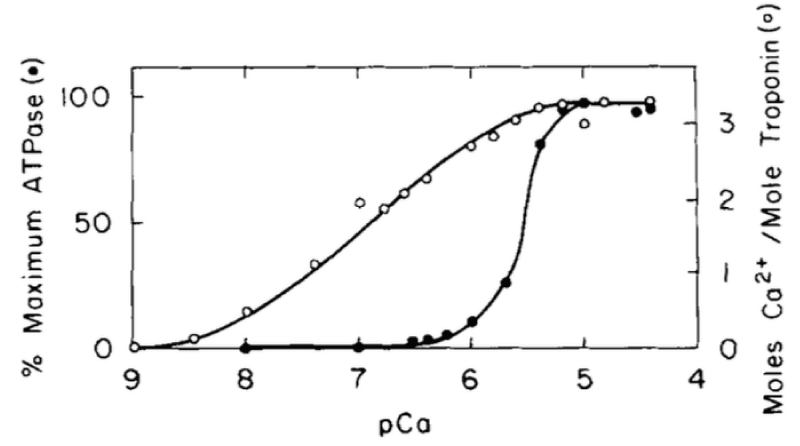
Figure 2: Comparison of Ca2+ activation of the cardiac myofibrillar ATPase and Ca2+ binding to cardiac (cTnC) in the presence of 4 mM Mg2+. (○) and the dependence of the myofibrillar ATPase activity on free (Ca2+) (●), from Holroyde, [1].
On cardiac cTnC there are two high affinity and two low affinity Ca2+ binding sites but only a total of three bind, i.e. only one for the weaker, activating sites. The strong binding sites are fully occupied under all physiological conditions. Mg2+ binding to any cTnC site is too weak to have the effect seen later under Magnesium inhibition. The conclusion is under physiological conditions the measurable binding of Ca2+ to the myofibrils is unimolecular, not cooperative.
This is fully confirmed by Morimoto and Ohsuki [3]. Only one of the low active sites is used on activation.
The competitive inhibition of Ca2+ activation by Mg2+ (Table 1).
| Table 1: Variation with pMg of the pCa50 (half activation) and Hill coefficient for Ca2+ activation of isometric force development in skinned cardiac-muscle fibres, from Smith, et al. [4] and original data from Donaldson [5]. | ||||
| pMg | 4.3 | 3.0 | 2.3 | 2.0 |
| pCa (half activation) | 5.56 | 5.06 | 4.59 | 4.37 |
| Hill coefficient (n) | 2.92 | 1.89 | 1.86 | 1.62 |
The conclusion is that activation is by Ca2+ bound to cTnC cooperatively with Ca2+ bound to the ATP on the myosin light chain in equilibrium with inactive myosin bound MgATP. Mg2+ does not competitively bind to cTnC. This ensures that the initial binding of MgATP in diastole gives the completely relaxed myofibril maintaining the status quo of the stress-growth equilibrium. Ca2+ is replaced by Mg2+ on cleavage of the pyrophosphate bond in ATP. MgATP bound to myosin light chain is derived from creatine kinase rephosphorylation of the myosin bound MgADP product of the cross-bridge ATPase. Creatine kinase is piggy-back bound to cardiac myosin binding protein-C (cMyBP-C) on the thick filament.
cMyBP-C bound to Actin-Myosin is required to maintain Ca2+ cooperativity (Figure 3).
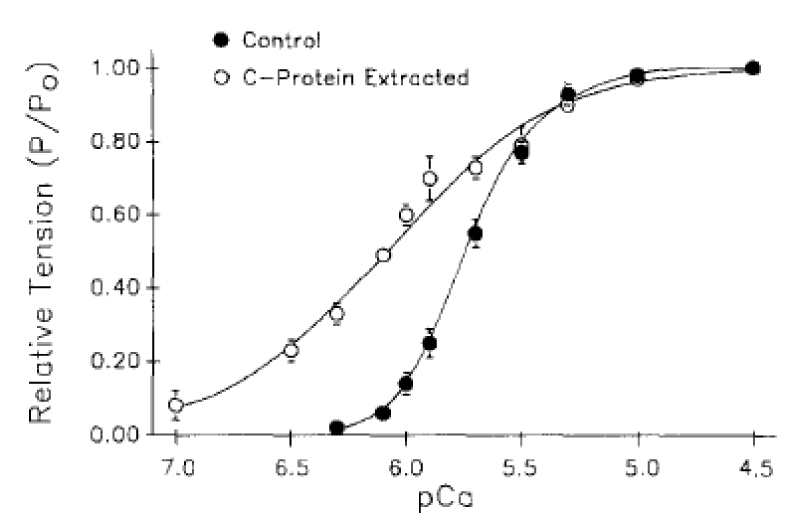
Figure 3: Tension-pCa data obtained before and after partial extraction of cMyBP-C (C-protein) from rat ventricular myocytes, from Hofmann, et al. [6], see cMyBP-C-nul later.
(a) The reversible extraction of cMyBP-C.
The displacement is to the left, lower [Ca2+] approaching the Km for cTnC Ca2+ binding (Table 2). The Ca2+ cooperativity is lost, lower Hill coeficient (nH), CaATP bound is not required. This demonstrates that MgATP can act as substrate when cMyBP-C is not bound to myosin-actin.
| Table 2: The increase in pCa50 and loss of cooperativity on removal of cMyBP-C. | ||
| pCa50 | n1 (P/P0 > 0.50) | |
| control | 5.77±0.03 | 2.22±0.19 |
| cMyBP-C extracted | 6.13±0.03 | 1.20±0.16 |
The process of removal is reversible (Figure 4).
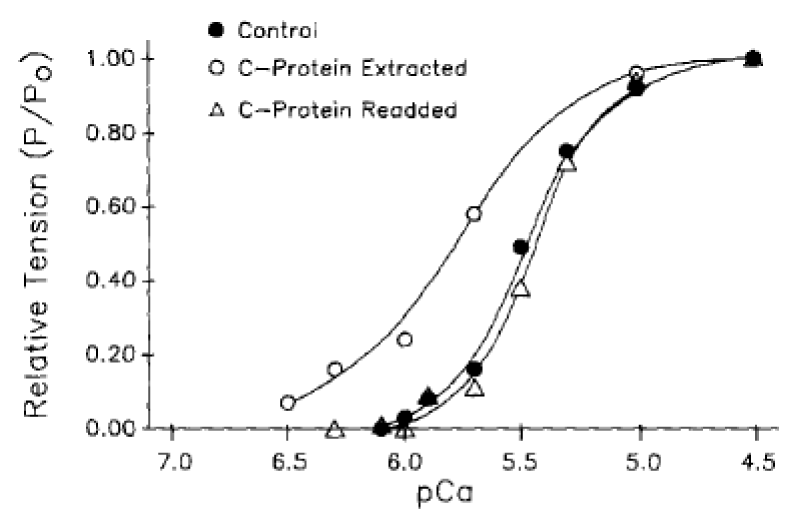
Figure 4: Tension-pCa relationships demonstrating the effects of rat cardiac C-protein readdition to a previously extracted cardiac myocyte, from Hofmann. et al. [6].
The conclusion is that cMyBP-C (along with cTnI see later) has the biochemical function of ensuring myosin bound MgATP is not the functional substrate of the cross-bridge ATPase in the normal heart. This ensures that the system is fully relaxed in diastole on completion of the cycle with binding of MgATP. cMyBP-C clearly has little structural function from the reversibility of its removal, save for its localisation of the creatine kinase.
(b) The addition of an N-terminal fragment of cMyBP-C that binds the myosin S2 site and actin.
Kampourakis, et al. [7] demonstrated the result of occupation of the free cMyBP-C binding sites (S2) on myosin by cMyBP-C myosin binding fragment C1mC2, see later. The free S2 binding sites outnumber those bound by a factor of at least 5. This N-terminal fragment of cMyBP-C contains the binding sites for myosin and actin binding. On addition this fragment binds to the free myosin and actin sites over-riding the effect of bound cMyBP-C [7]. At the concentration of C1mC2 used (2 µM) it almost certainly does not displace the bound cMyBP-C, as its Km for maximal Ca2+ free activation is more than 20 µM. This ability to fully activate in the absence of Ca2+ is the first indication that the action of cMyBP-C also involves the cTnI, i.e. is concerted (Figure 5).
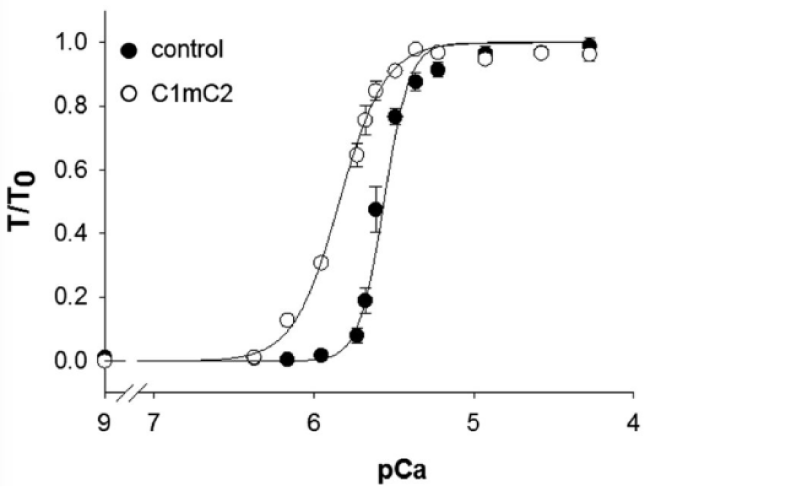
Figure 5: The effect of binding of cMyBP-C fragment C1mC2 to acto-myosin, from Kampourakis, et al. [7].
The displacement is to the left, lower [Ca2+], and the cooperativity is lost. Only Ca2+ bound to cTnC is required for activation, therefore MgATP can act as substrate when C1mC2 is bound to myosin-actin. cMyBP-C bound to Myosin and/or the presence of free Myosin S2 sites are required to maintain Ca2+ cooperativity.
The stress/strain relationship to cardiomyopathies
I have previously suggested [8] that any mutation giving rise to the above loss of Ca2+ cooperativity would mean that true diastole would not be reached and the resulting chronic strain would be transmitted to the sensor in titin, releasing LIM protein and hence promoting growth. The result of increased growth is hypertrophic cardiomyopathy (HCM) [8]. Any reduction in transmission of tension to the titin would result in not maintaining the status quo of growth v. tissue loss, less Lim activity reduced maintenance growth and dilated cardiomyopathy (DCM). When mediated through change in the cMyBP-C resulting in HCM the shift in sensitivity is to the left (lower Ca2+) and the opposite for DCM.
Sarcomeric mutations giving rise to a reduction in the Ca2+ cooperativity of activation giving rise to HCM
Disruption of either the cTnI or the cMyBP-C functions results in the myosin bound MgATP being used as substrate, with loss of calcium cooperativity and a shift to increased Ca2+ sensitivity. In general the result of this is incomplete relaxation at the end of the cross-bridge cycle when MgATP is rebound and immediately used when some cTnC at diastolic calcium level ([Ca2+]D) is still Ca2+ bound, causing chronic tension in the myofibril. This tension is transmitted through the troponin-actin-titin system, with release of growth activators (LIM protein) and resulting hypertrophic growth of the myocytes.
HCM has been recently reviewed by Teekakirikul, [9]. Along with DCM it is one of the most common heritable cardiovascular disorders. HCM is characterized by left ventricular hypertrophy (LVH) that is unexplained by abnormal loading conditions, with myocyte hypertrophy and disarray, and increased myocardial fibrosis as key histopathological hallmarks. Genetic studies reveal the multiple variants in sarcomere protein genes in approximately 40% – 60% of patients with HCM, establishing HCM as a disease of sarcomere proteins, similarly for DCM. Most HCM disease-causing variants occur in the myosin, cMyBP-C and cTnI. HCM variants do also occur, along with other myopathy causing mutations, in the thin filament proteins.
I now give examples of instances of HCM where the kinetics have been graphically reported. Most of these show a small consistent loss of Ca2+ cooperativity reflected in reduced Hill coefficient (nH) for Ca2+ activation, accompanied by increased Ca2+ sensitivity.
Mutations in cMyBP-C giving HCM (Figure 6).
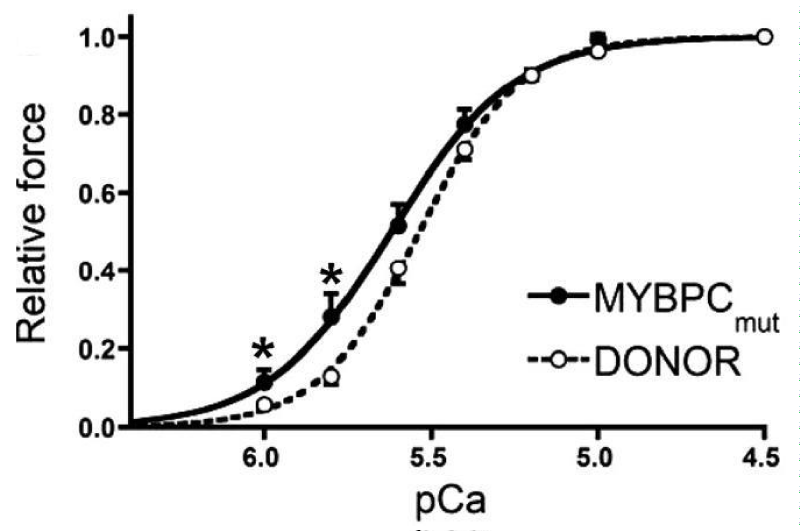
Figure 6: Tension pCa relationship for frameshift mutants of cMyBP-C, from van Dijk, et al. [10].
Along with Myosin mutations these are the main sources of HCM and here the effect of cMyBP-C knockout is included.
(a) cMyBP-C mutant carriers c.2373dupG and 2864_2865DelCT.
The mutant cMyBP-C showed a shift to lower [Ca2+] and reduced cooperativity. Expression of cMyBP-C was also reduced by 30% (Figure 7).
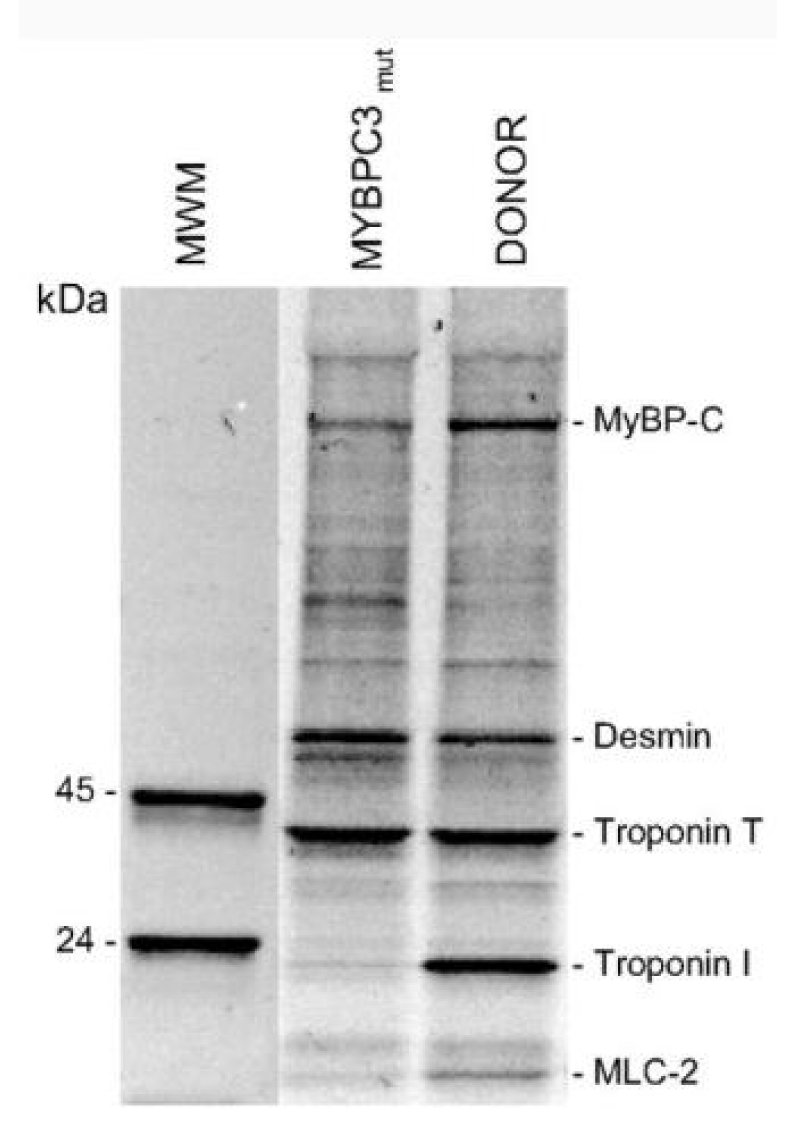
Figure 7: The levels of phosphorylated proteins. van Dijk, et al. [10].
Here they have also shown that for this mutation of cMyBP-C there is almost complete absence of cTnI phosphorylation compared to normal. This appears to be accompanied with a decrease in phosphoryl cMyBP-C, more than the protein level decrease, and an increase in that of desmin [10]. This is the second strong link in the activities of cMyBP-C and cTnI.
(b) KOY235S mutation of cMyBP-C (Figure 8).
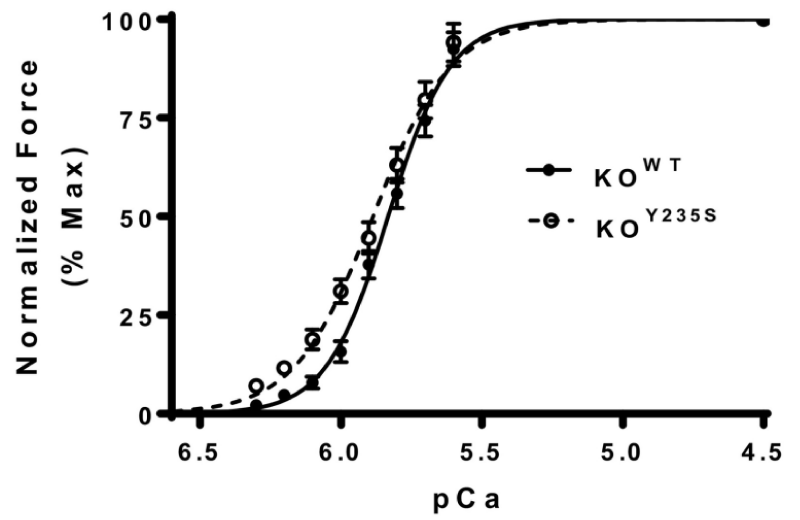
Figure 8: Force-pCa relationship between KOWT and KOY235S of cMyBP-C, from Doh, et al. [11].
The KOY235S myocardium shows a left-ward shift in the pCa curve, indicating increased calcium sensitivity and reduced Ca2+ cooperativity. Unlike the frameshift mutants above there is no accompanying reduction in cMyBP-C expression or phosphorylation of it or the cTnI.
Parbhudayal, et al. [12] have also made the first study to demonstrate an intercellular variation of myofilament cMyBP-C protein expression within the myocardium from HCM patients with heterozygous cMyBP-C mutations.
(c) cMyBP-C knockout mice.
Some of the observations on the genetic elimination of the cMyBP-C are contrary to those on the extraction of cMyBP-C [6] or binding of C1C2 [7] fragment. These studies used gene targeting to produce knockout mice that lack cMyBP-C in heart. The results show that cMyBP-C is not essential for sarcomere assembly or cardiac development but that the absence of cMyBP-C is sufficient to trigger profound cardiac hypertrophy and depressed myocyte contractile properties. Clearly in the absence of the substrate control by cMyBP-C one would expect both an increase in Ca2+ sensitivity and loss of cooperativity. However, the finding that Ca2+ sensitivity of tension was reduced in cMyBP-C-nul mice contrasts with enhanced Ca2+ sensitivity reported after extraction of ∼60% of cMyBP-C from rat myocytes using biochemical techniques. Potential explanation to account for the different results have been suggested. These are that the biochemical extractions occurred in vitro over short time periods, thus precluding adaptive responses, whereas compensatory effects (eg, protein phosphorylations or structural changes during hypertrophy) after genetic elimination of cMyBP-C may be additional factors in the present study. However a better explanation is in binding of the cMyBP-C to the actin-myosin, see effect of N-terminal fragments later.
The first study of cMyBP-C-nul By Harris, et al (Figure 9).
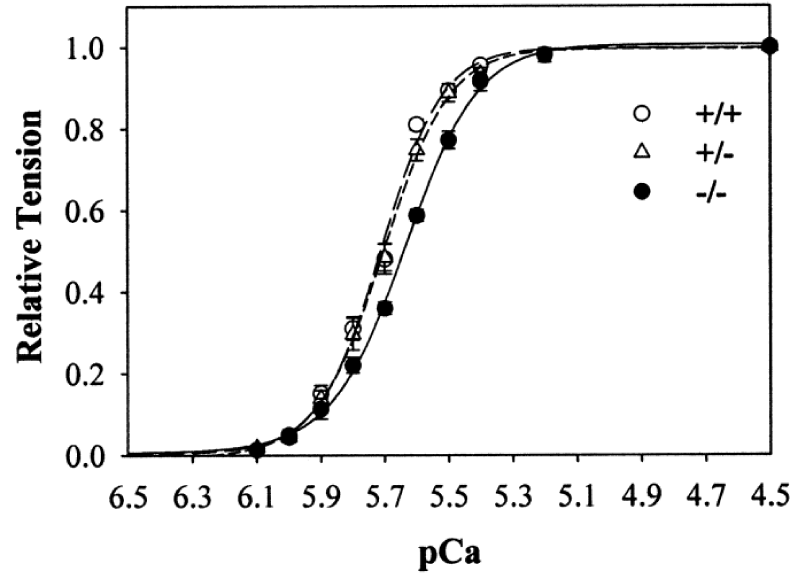
Figure 9: Ca2+ sensitivity of tension in skinned myocytes from cMyBP-C-nul mice. wild-type (+/+), heterozygous (+/−), and homozygous (−/−) knockout, from Harris, et al. [13].
Here the anomalies found were a less than expected reduction in the Ca2+ cooperativity and much less than expected Ca2+ sensitivity, cf ref [6,7].
In a follow up Harris, et al. found [14] a more expected effect of the cMyBP-C knockout on the Ca2+ cooperativity, cf Hofmann [6]. But retained the lower sensitivity. In this study they surprisingly looked at the effects of adding binding fragments a la Kampourakis, et al. [7] (Figures 10-12).
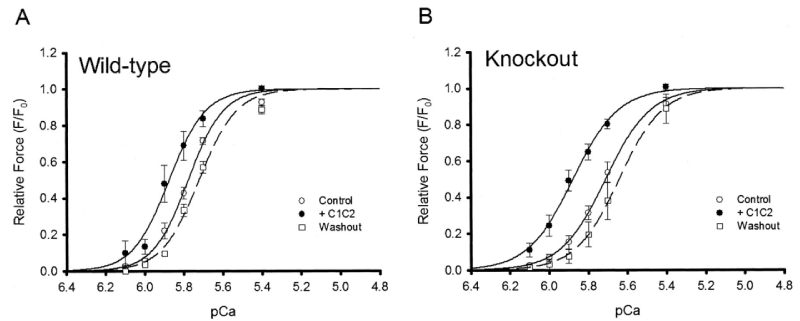
Figure 10: Effect of C1C2 peptide (10 µM) on tension-pCa relationships in permeabilized myocytes, Wild type and cMyBP-C-nul knockout, from Harris, et al. [14].
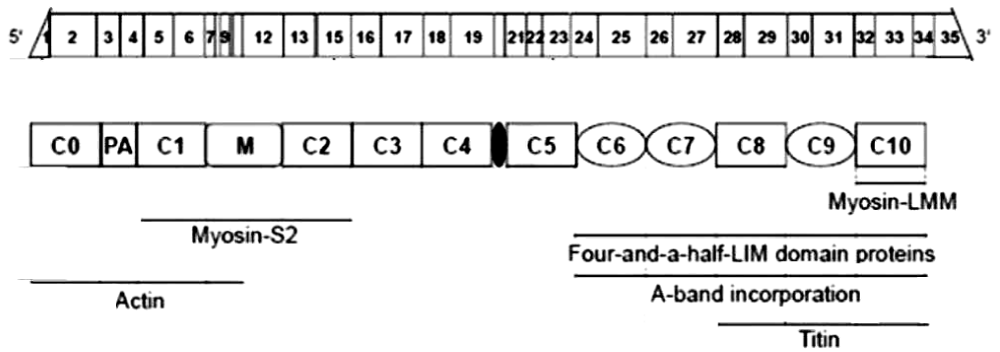
Figure 11: Schematic representation of cMyBP-C mRNA and protein structure and its interaction partners from Carrier, et al. [16].
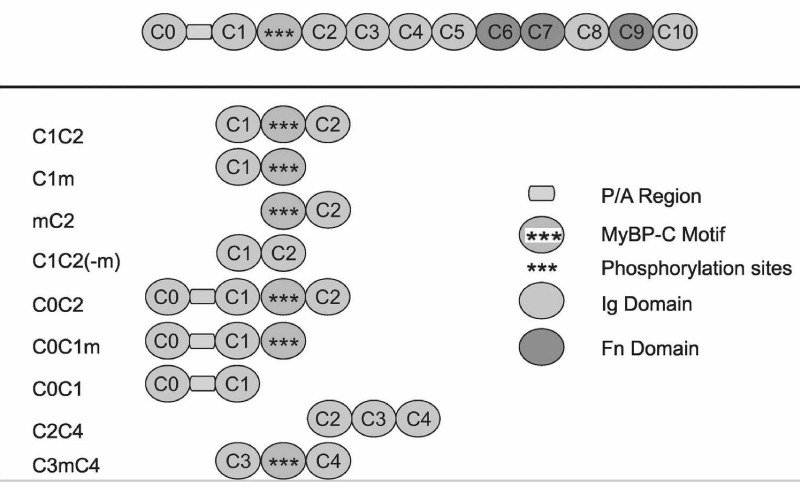
Figure 12: Modular Domain Organisation of Cardiac cMyBP-C and the fragments synthesised [15].
Of note is the change of pCa50 with lack of reduction in Hill coefficient on addition of C1C2 fragment to the wild-type in this later study [14] in contrast to that of others, Kampourakis, [7]. Figure 5 and Razumova [15], Figure 13. The shift to greater Ca2+ sensitivity on addition of C1C2 in the absence of cMyBP-C confirms the effects of C1C2 fragment are by direct binding to unoccupied sites on the myosin (S2) and/or the actin, as suspected from the concentration used by Kampourakis. et al. [7] and the concentration they required for Ca2+ free activation. The dose dependency of C1C2 compared to that of C0C2 or C0C1m on the cMyBP-C KO would be interesting in this regard. The lack of shift to lower Ca2+ by knockout indicates a weaker affinity of the cTnC for Ca2+ in the absence of cMyBP-C, i.e. cMyBP-C binding to the actin-myosin increases the affinity of cTnC for Ca2+. The large leftward shift to greater Ca2+ sensitivity on addition of C1C2 fragment in the KO case does confirm this. Only partial removal of cMyBP-C, Hofmann, [6], leaves some cTnC with the higher affinity.
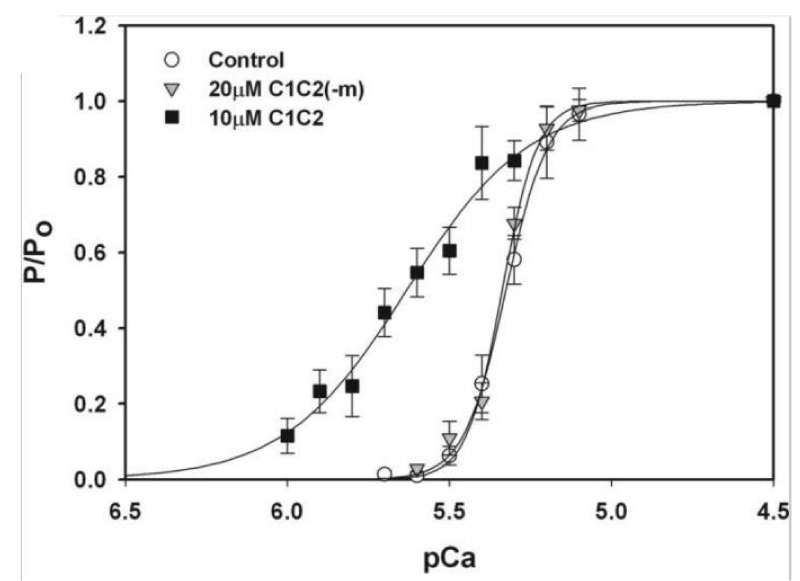
Figure 13: Effects of the cMyBP-C motif on Ca2+ sensitivity of tension, from Razumova, [15].
Limitations on use of cMyBP-C fragments.
For convenience I include here the mapping of the N-terminal fragments of cMyBP-C (Figures 11,12).
Incubation of trabeculae with 10 μM C1C2 resulted in a leftward shift of the tension–pCa relationship relative to control, indicating an increase in Ca2+ sensitivity of tension (ΔpCa50 = 0.30 ± 0.05). This confirms the incorrectness of the observation of Harris, et al. [14] (Table 3).
| Table 3: The effects of N-terminal proteins on Ca2+ activation [16]. | ||||||
| Protein (n) | [ ] (µM) | pCa50 | pCa50 | ∆pCa50 | Hill coef. (nH) | Hill coef. (nH) |
| control | + protein | control | + protein | |||
| C0C2(6) | 5 | 5.32±0.02 | 5.73±0.04 | 0.42±0.04 | 5.43±0.32 | 2.19±0.14 |
| C1C2(4) | 5 | 5.30±0.02 | 5.54±0.05 | 0.25±0.05 | 6.45±0.86 | 2.65±0.25 |
| C0C1m(4) | 5 | 5.32±0.01 | 5.59±0.05 | 0.27±0.05 | 7.66±0.72 | 2.71±0.34 |
| C1m(6) | 5 | 5.34±0.02 | 5.46±0.04 | 0.12±0.04 | 6.83±0.89 | 3.72±0.34 |
| C1C2(5) | 10 | 5.29±0.02 | 5.58±0.05 | 0.30±0.05 | 5.02±0.33 | 2.11±0.14 |
| mC2(4) | 10 | 5.30±0.01 | 5.32±0.03 | 0.02±0.03 | 6.63±0.44 | 5.90±0.67 |
| C0C1(3) | 10 | 5.34±0.01 | 5.32±0.01 | -0.02±0.01 | 6.61±0.70 | 7.50±0.83 |
| C2C4(4) | 20 | 5.32±0.01 | 5.32±0.01 | 0.00±0.01 | 6.73±0.21 | 6.15±0.71 |
| C1C2m(3) | 20 | 5.33±0.02 | 5.34±0.01 | 0.01±0.01 | 9.62±2.08 | 8.67±2.04 |
| C3mC4(3) | 20 | 5.30±0.02 | 5.29±0.01 | -0.01±0.01 | 4.96±0.62 | 5.56±0.42 |
The addition of 20 μM C1C2(-m) that lacks the cMyBP-C motif was without effect on the tension–pCa relationship.
Note the use of only 5 µM of the N-terminal domain peptides. Km for C1C2 Ca2+ free activation is 20 µM [7], when m is phosphorylated C1C2 becomes inactive. Kampourakis, et al. [7] only used 2 µM so minimising Ca2+ free stimulation. In experiments with cMyBP-C nulls Raumova, et al. [16] use 10 µM. The 20 µM Km for maximum activation without Ca2+ probably reflects displacement of bound cMyBP-C, 2 µM only binds at free Myosin S2 and or actin sites. Hence the large reduction of Hill-coeficient on addition.
The much later observations of Harris, et al. [17] have been ignored as.
They used 100 µM of the fragments and in doing so even C1-m was found to act alone.
Mutation in myosin light chain resulting in HCM (Figure 14).
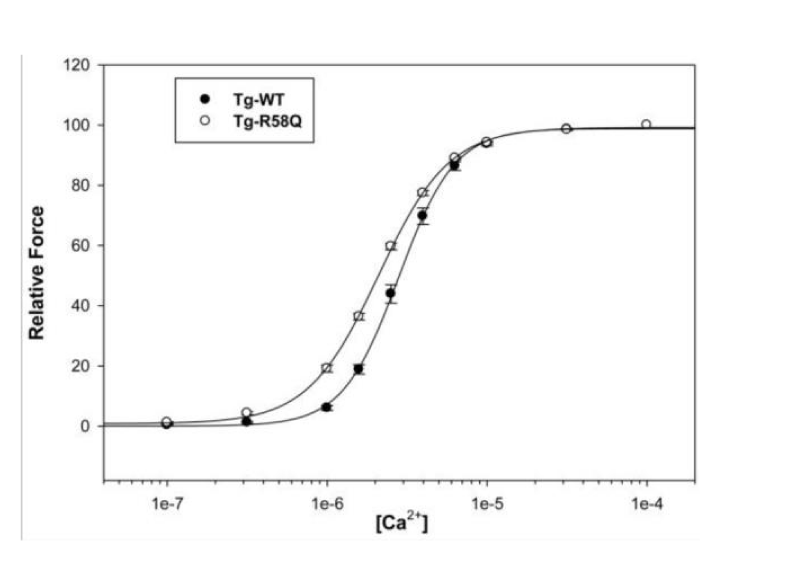
Figure 14: The effect on Ca2+ sensitivity of force determined in Tg-R58Q glycerinated skinned papillary muscle fibers compared to Tg-WT, from Mettikolla, et al. [18].
The shift to lower [Ca2+] and loss of cooperativity was accompanied by considerable loss of contraction strength, however this does bypass the cMyBP-C substrate control and the linkage is not floppy as with DCM mutations in light chain, see later.
Mutation in cTnI resulting in HCM (Figure 15).
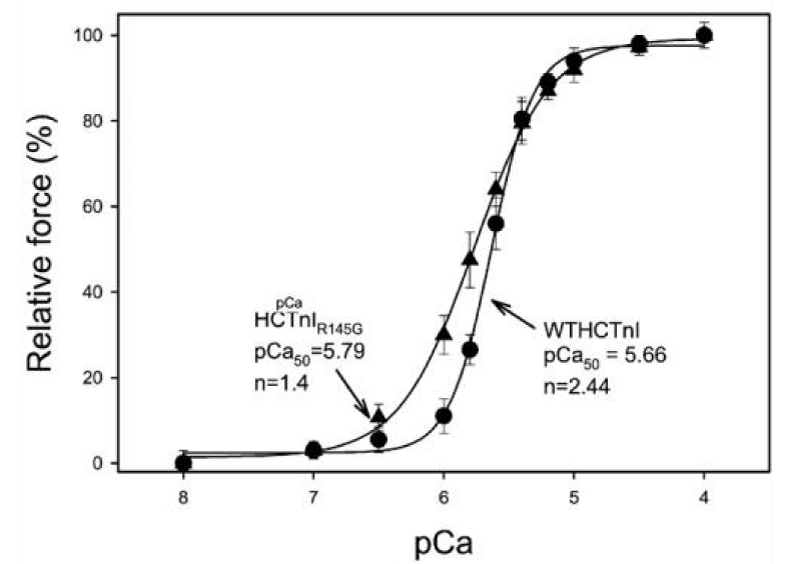
Figure 15: Activation of actin-Tm-activated myosin ATPase activity by either WTHCTnI or HCTnIR145G, dependence on Ca2+, from Lang, et al. [19].
The observed results of this mutation were;
1) the complex containing HCTnIR145G only inhibited 84% of Ca2+-unregulated force. Incomplete relaxation at diastolic Ca2+, chronic stress.
2) the recovery of Ca2+-activated force by HCTnIR145G was decreased and
3) there was a significant increase in the Ca2+ sensitivity of force development.
Restrictive cardiomyopathy (RCM) with cTnI mutations.
There are reports of restrictive cardiomyopathy (RCM) showing major loss of Ca2+ cooperativity arising from mutations mostly in cTnI. RCM is a more pronounced form of HCM. Two examples are given here, both from Davis, et al. (Figures 16,17).
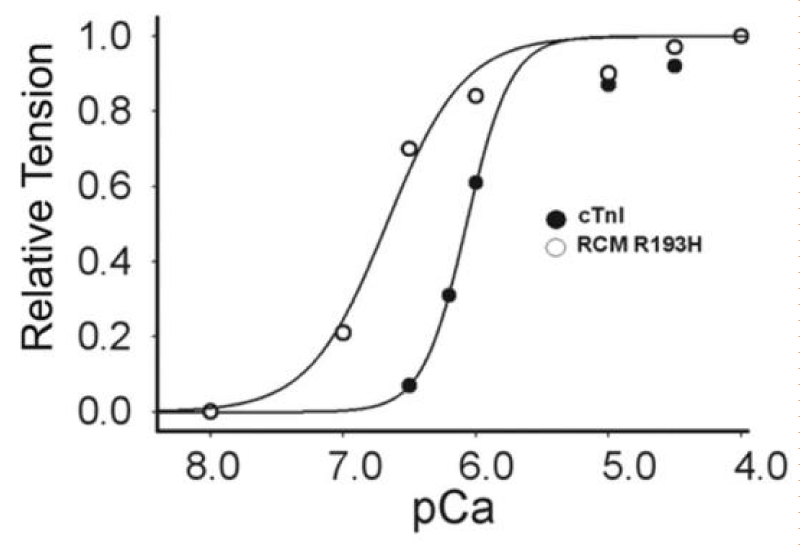
Figure 16: Ca2+-activated tension in single permeabilized R193H cTnI mutant cardiac myocytes, from Davis, et al. [20].
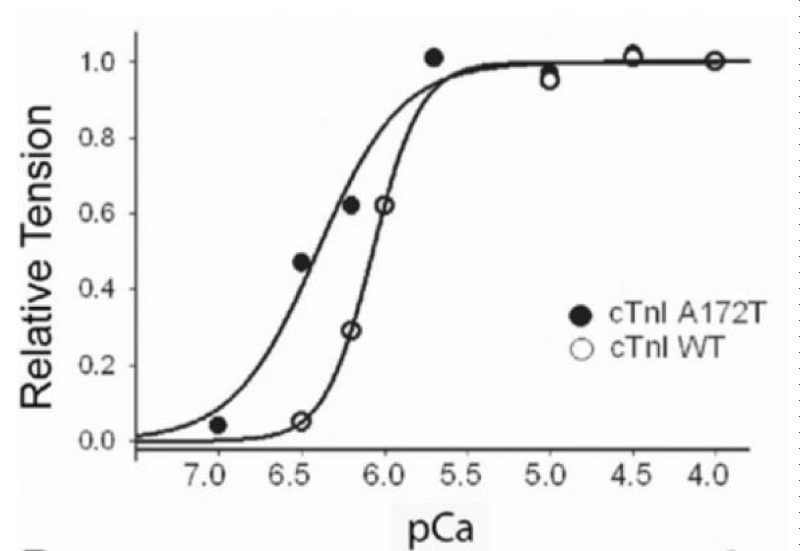
Figure 17: Ca2+-activated tension in single permeabilized A172T cTnI mutant cardiac myocytes, from Davis, et al. [21].
PKA treatment shifts both curves to the right, less Ca2+ sensitive without change in Hill coefficients.
RCM is when the walls of the lower chambers of the ventricles are too rigid to expand as they fill with blood. The pumping ability of the ventricles may be normal, but it’s harder for the ventricles to get enough blood. With time, the heart can’t pump properly. This leads to heart failure. With RCM mutants the loss of cooperativity and shift to greater Ca2+ sensitivity are the largest of those reported for HCM and more reflect those found when cMyBP-C is nullified by addition of its S2 binding moiety or partially removed. There is considerable activation and lack of relaxation at low [Ca2+]D [20,21].
There is no evidence of amyloidosis giving rigidity in this case, as shown by Du, et al. [22], however increases in desmin and α-actinin have been demonstrated in diastolic failure by Zhang, [23] and Sheng, et al. [24]. The latter may very well be the basis of the increase in rigidity found in RCM.
Other RCM related cTnI changes are reported by Parvatiyar, et al. [25], Jean-Charles, et al. [26], Wen, et al. [27] and Gomes, [28].
With the above large shift to increased Ca2+ sensitivity the suggestion is that cTnI is involved with the binding of cMyBP-C to the actin-myosin, the third strong indication of this.
A mutation cTnC giving unexpectedly DCM although loss of Ca2+ cooperativity.
There is one instance of mutated cTnC where the use of MgATP as substrate will occur and should lead to hypertrophy but this is over-ridden by breakdown in stress transfer to the thin filament causing dilated myopathy (Figure 18).
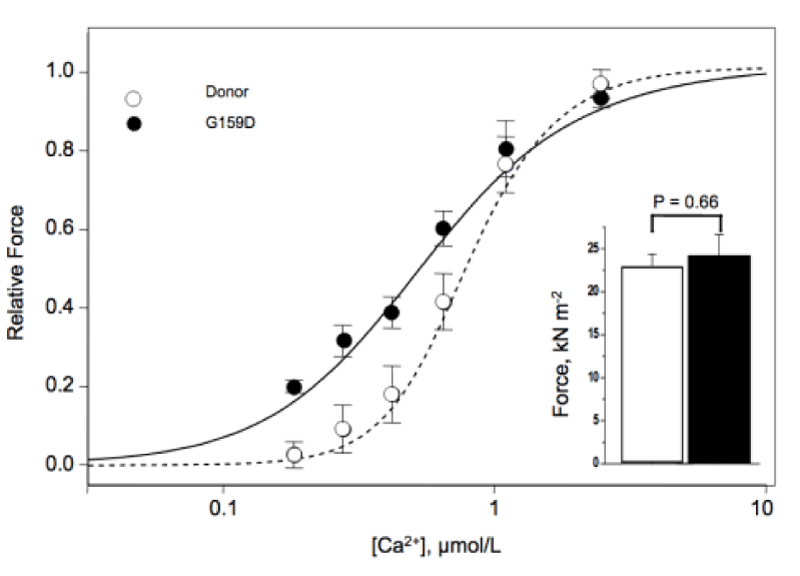
Figure 18: Contractile properties of skinned donor and G159D cTnC myocytes Inset, Maximum force in skinned myocytes from human donor (open bar) and G159C cTnC tissue (filled bar), from Dyer, et al. [29].
I conclude here that the Ca2+ sensitivity increase is entirely down to a structural change i.e. cTnI not being G159C-cTnC bound [27] and thus not cooperating with the cMyBP-C in blocking the use of MgATP as substrate. It is also likely that G159C-cTnC does not properly bind other thin filament proteins, actin, cTnT or tropomyosin. Similar results were found by Swindle, et al. [30].
Sarcomeric mutations that do not show loss of Hill coefficient, Ca2+ cooperativity, giving rise to either HCM or DCM
These mutations are mostly in the Troponin-T-tropomyosin-titin genes although those in myosin are reported. The mutations either increase or decrease the transfer of stress to the titin and its bound Lim protein, giving rise to HCM or DCM. The Ca2+ cooperativity is maintained, i.e. both the cTnC and myosin bound ATP are Ca2+ bound with these mutations. The central roles of Troponin-T (cTnT) and tropomyosin (∝-Tm) in the thin filament transfer of stress is well demonstrated by the presence of assorted variants with either hypertrophic or dilated outcome. The HCM mutants of ∝-tropomyosin (∝-Tm) all display elevated Ca2+ free ATPase (or tension), a form of inhibited diastolic relaxation, different to loss of Ca2+ cooperativity, again giving chronic stress in the titin. In general the various mutants giving DCM show reduced maximum ATPase/tension, little or no shift in either the pCa50 or Hill coefficient.
Mutations in the Myosin Heavy Chain
For the myosin heavy chain variants there are very little data on the variation of ATPase or contractility with [Ca2+] although there are a very large number of mutations known. All available are HCM and show increased maximum contractility at high [Ca2+]. The earliest found was Keller, et al. [31] who showed that mutant myosin R403W exhibited a large increase in maximal actin-activated ATPase activity (+114%; p < 0.05) and Km for actin (+87%; p < 0.05) when compared to WT. Kraft, et al. [32] found increase in contractile strength for R723G in cardiomyocytes. Spudich, et al. [33] studied the mutations R21C, S166F (small nH increase) and DK177 all showed increase maximal ATPase. Nag, et al. [34] for HCM R403Q Show a lowering of contractile strength. It lacks the S2 fragment (cMyBP-C binding domain) and thus must surely behave like cMyBP-C-nul. Sarkar, et al. [35] confirm the R403Q conclusions. These all increase tension transfer to titin.
Mutation in myosin light chain giving DCM (Figure 19)
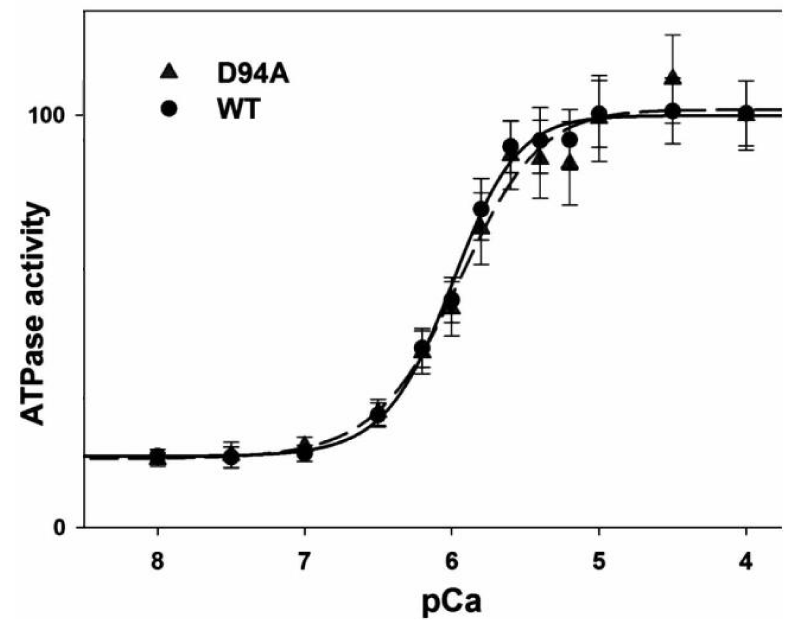
Figure 19: The ATPase-pCa relationship in WT and D94A reconstituted porcine cardiac myofibrils, from Huang, et al. [36].
This light-chain mutation was seen to impair the binding of the regulatory light chain (RLC) to the myosin heavy chain (MHC). Less binding to MHC, more floppy less strain is transferred to the thin filament.
Tropomyosin (∝-Tm) mutations giving HCM
Michele, et al. [37] report the functional effects of cardiac-specific expression of human E180G mutant ∝-Tm in transgenic mice (Figure 20).
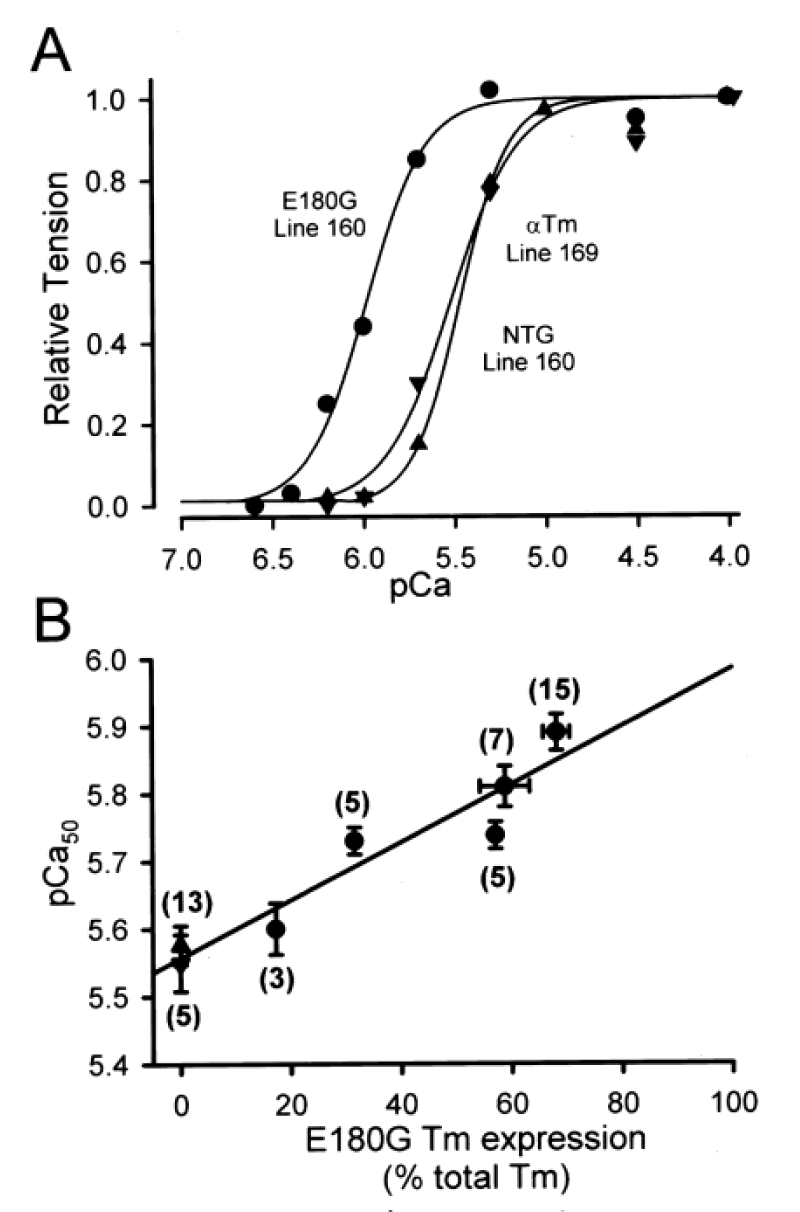
Figure 20: E180G ∝-Tm transgene dose dependence of increased Ca2+ sensitivity of force production in single cardiac myocytes. A, Ca2+-activated force generation in representative single cardiac myocytes isolated from NTG (▲), 169 WT ∝-Tm (▼), and 160 E180G ∝-Tm (●) mice. B, E180G ∝-Tm transgene dose dependence of increased Ca2+ sensitivity of force production, from Michele, et al. [37].
E180G ∝-Tm mice had significantly slowed relaxation under physiological conditions. This dysfunction was eliminated by propranolol. In a follow up by Michele, et al. [38] they use adenoviral-mediated gene transfer of four point mutations in α-tropomyosin (∝-Tm) into adult cardiac myocytes in vitro to show that all four HCM α-Tm proteins can be expressed and incorporated into normal sarcomeric structures in cardiac myocytes at similar levels as normal α-Tm proteins.
Muthhuchamy, [39] report the functional effects of cardiac-specific expression of HCM human α-TM 175 TG in transgenic mice (Figure 21).
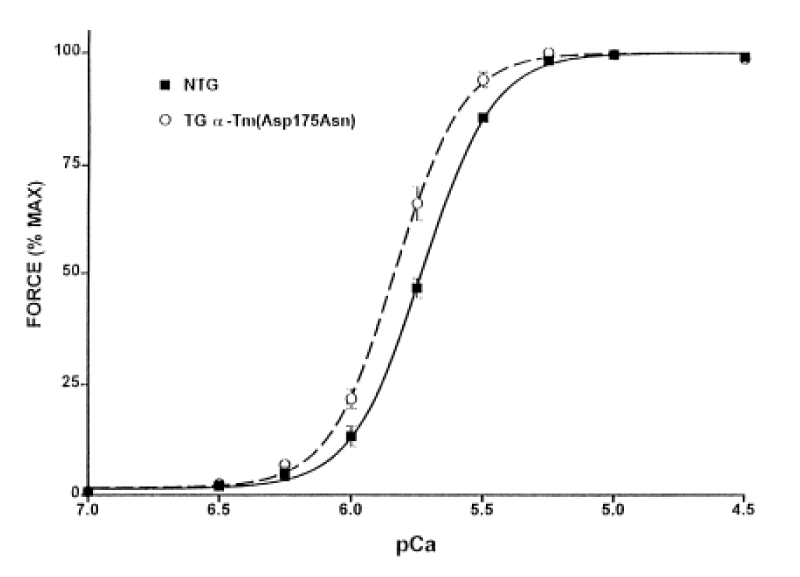
Figure 21: Normalized pCa-force relation of skinned fibre bundle preparations from NTG and α-TM 175 TG (line 108) mouse hearts at sarcomere length 2.0 μm.
Increased expression of the α-TM 175 transgene in different lines causes a concomitant decrease in levels of endogenous α-TM mRNA and protein expression. In vivo physiological analyses show a severe impairment of relaxation in hearts of the HCM mice. In this case the stress was elevated in diastole. Chang, et al. [40] report similar results with other HCM-associated ∝-Tm mutations (Figure 22).
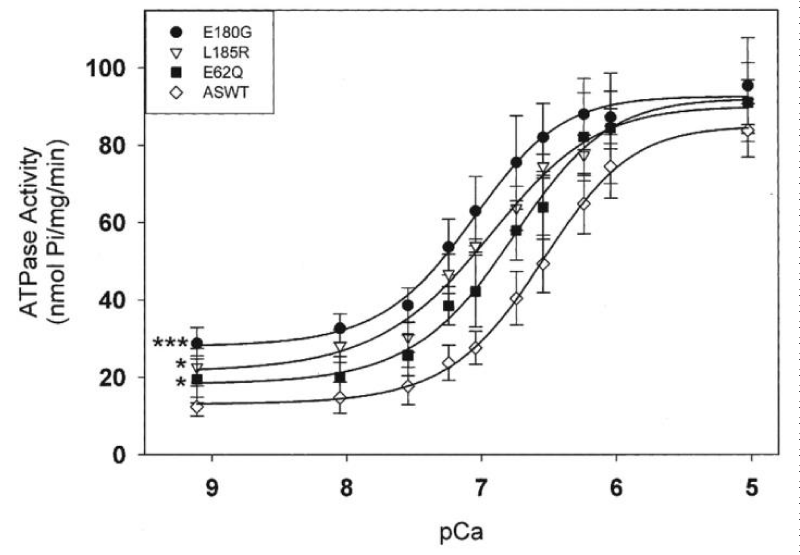
Figure 22: Effects of HCM-causing ∝-Tm mutations on the Ca2+-dependent myofibrillar ATPase activity rates. Chang, et al. [40].
All three HCM-associated ∝-Tm mutations increased the Ca2+ sensitivity of ATPase activity. All three had significantly decreased abilities to inhibit ATPase activity at effectively zero [Ca2+]. This is clearly the same as with the α-TM 175 transgenic above [39]. In all these the [Ca2+]D chronic stress is transferred to the titin.
∝-Tm mutants giving DCM (Figure 23).
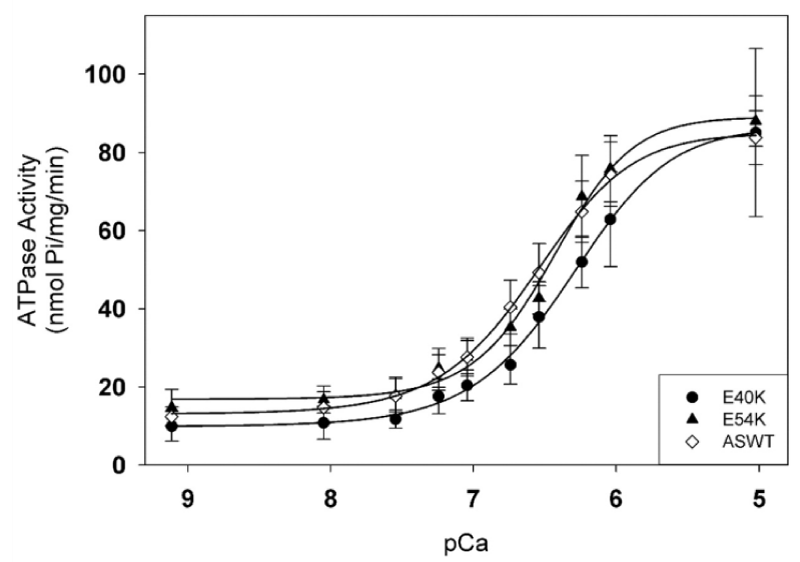
Figure 23: Effects of DCM-causing ∝-Tm mutations, E40K and E54K, on Ca2+-dependent myofibrillar activity rates compared to WT.
Statistical analysis of activities at pCa 9.1 and 5.0 shows no significant differences; Both mutations caused a small but significant decrease in the pCa50 values, and had no effect on the inhibition of ATPase activity. from Chang, et al. [40]. Somehow less tension in titin, need to look for altered interaction with cTnT etc.
Various reports of other HCM and DCM Tm mutants
Gupte, et al. [41] showed DCM mutants D84N and D230N shift the pCa50 stimulated ATPase activity to the right relative to WT and HCM mutants shift the pCa50 to the left. The changes in pCa, nH and ATPasemax for the DCM cases are small. The changes in the curves for the HCM mutants are again similar to above ∝-Tm reports and there is residual activity at diastolic [Ca2+]D. Similar DCM results are reported by Rajan, et al. [42] for a-TM54 TG hearts with more significant loss in contraction force (Table 4) (Figure 24).
| Table 4: Ca2+-tension in skinned fibre bundles of a-TM54 TG hearts [42], DCM. | |||
| Group | pCa50 | nH | Force (mN/mm2) |
| NTG (5 months old) | 5,75±0.02 | 3.38±0.40 | 37.50±4.04 |
| Moderate copy TG | 5.67±0.01 | 3.77±0.21 | 25.41±1.57 |
| NTG (1 month old) | 5.93±0.03 | 3,74±0.23 | 46.09±7.22 |
| High copy TG, 1.9 | 5.82±0.03 | 3.78±0.15 | 22.35±1.48 |
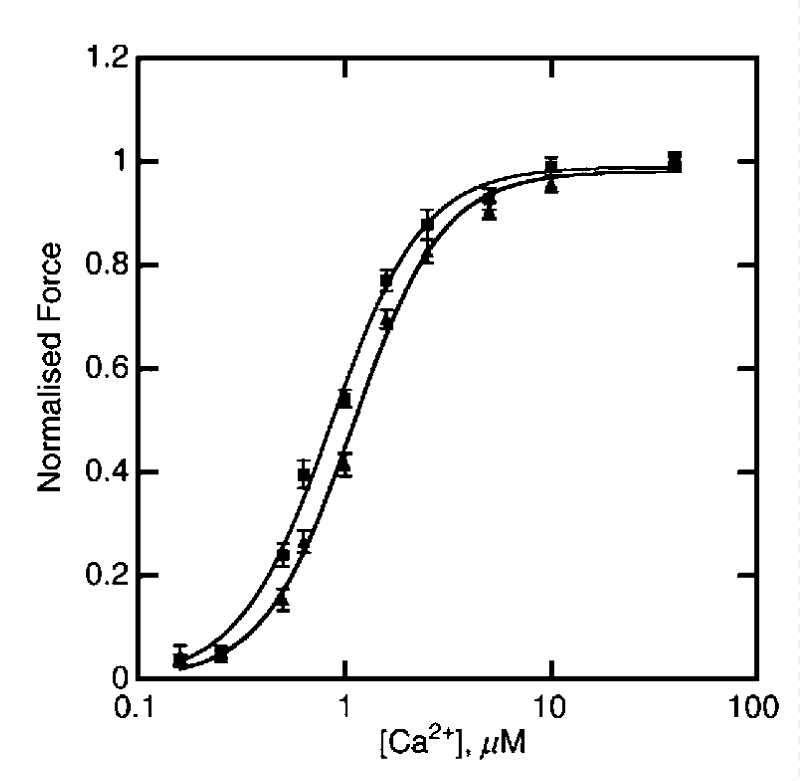
Figure 24: The dependence of isometric force on [Ca2+] for 8 NTG(▲) or ACTC E361G mice (■), from Song, et al. [43].
Actin mutant giving DCM
There is no cooperativity change and a small movement to lower [Ca2+]. There was no significant difference in sliding speed or fraction of filaments motile. When troponin was dephosphorylated the Ca2+-sensitivity of E361G-containing thin filaments was now much lower than NTG, this was due to uncoupling of Ca2+-sensitivity from cTnI phosphorylation.
cTnT mutations giving HCM
Hernandez, et al. [44] report both ATPase and force measurements for skinned papillary muscle fibers of F110I-cTnT and R278C-TnT transgenic mice. In both F110I-TnT and R278C-TnT the maximum force was greatly reduced from WT but the ATPase not affected. In this case it is clear that the force instead of transmitting to the Z disc was absorbed by the titin spring, and thus releasing increased LIM protein.
cTnT mutations giving both HCM and DCM
(a) human wild-type, ΔK210 (DCM), or ΔE160 (HCM) cTnT (Figures 25,26).
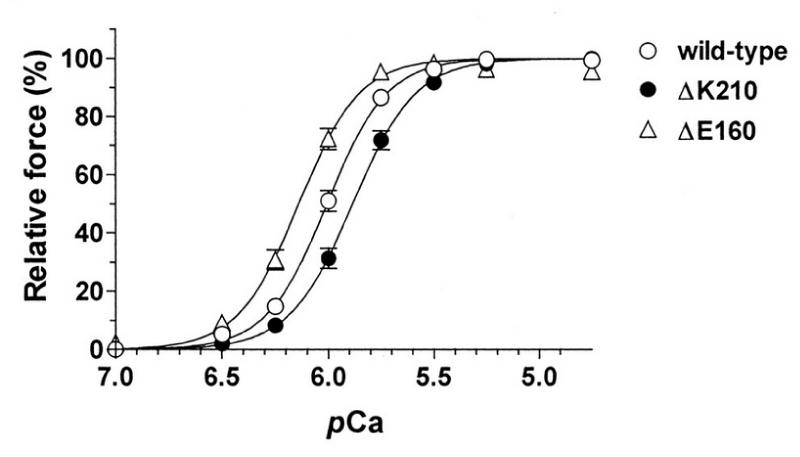
Figure 25: Force–pCa relationships in the rabbit skinned cardiac muscle fibers into which human wild-type, ΔK210 (DCM), or ΔE160 (HCM) cTnT was incorporated. Maximum force levels were both ca. 30% down compared with wild-type, from Morimoto, et al. [45].
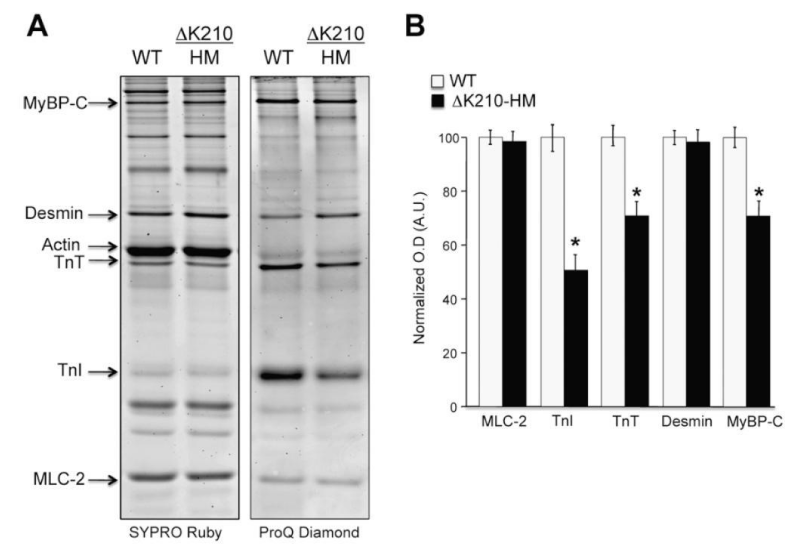
Figure 26: Differential phosphorylation of sarcomeric proteins in isolated myofibrils from ΔK210 homozygous hearts (DCM). A, cardiac myofibrillar proteins stained with ProQ Diamond for phospho-proteins detection and Sypro Ruby for total protein detection. They show equal protein content (Sypro) and disparity in phosphorylation levels (ProQ), from Sfichi-Duke, et al. [46].
Venkatraman, et al. [47] report similar results for other DCM mutants.
(b) HCM cTnT-I79N mutation (I79N). DCM knock-in mouse model carrying the heterozygous TnT-R141W mutation (HET) (Figure 27).
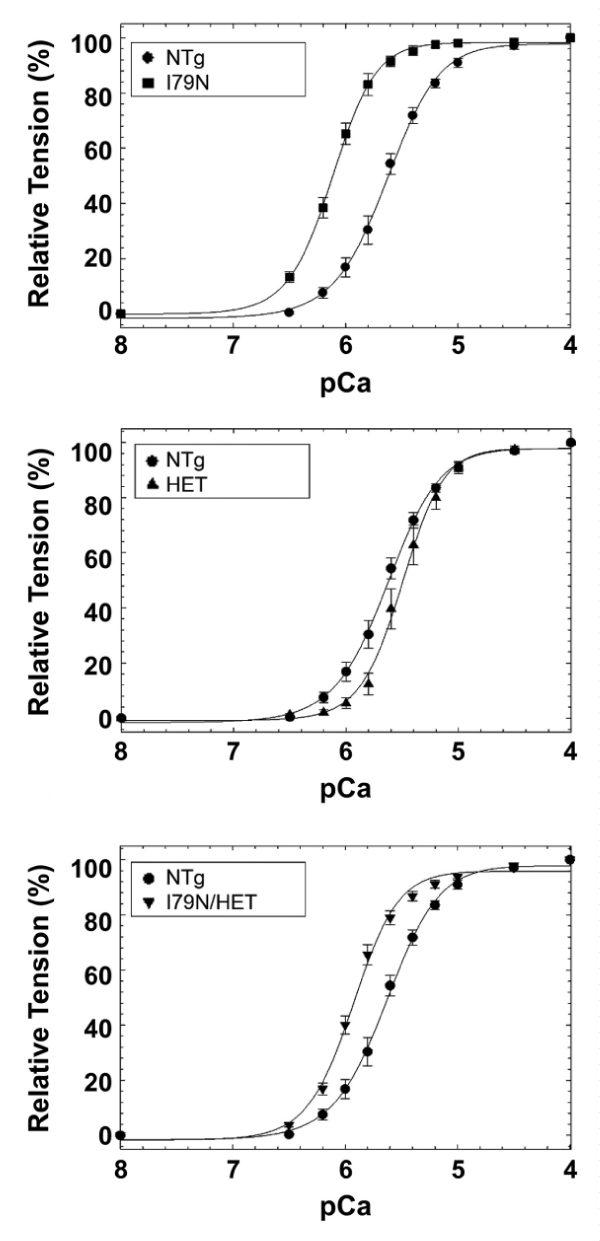
Figure 27: Calcium-dependent tension of, A, I79N mice compared to NTg. B, R141W HET mice compared to NTg. C, I79N/HET mice mutually normalized the Ca2+ sensitivity towards WT, from Jones, et al. [48].
It appears that the transfer of stress to the titin is opposite in the two cases and when both are included together they nullify each other. In this case there are small changes in Hill coefficient, origin of which is not clear.
Sommese, et al. [49] report on mutants used above (Table 5).
| Table 5: ATPase activity of cardiac S1 with assorted HCM and DCM TnT mutants [49]. | ||||||
| TnT | disease | pCa50 | ∆pCa50 | % max.activity | % min.activity | nH |
| WT | 6.17±0.01 | - | 100±1 | 10±1 | 1.2±0.1 | |
| 179N | HCM | 6.44±0.04 | +0.27 | 100±2 | 7±1 | 1.2±0.1 |
| E163K | HCM | 6.36±0.03 | +0.19 | 147±7 | 15±1 | 1.4±0.2 |
| R141W | DCM | 5.96±0.02 | -0.21 | 93±2 | 5±1 | 1.2±0.1 |
| R173W | DCM | 6.08±0.01 | -0.09 | 101±4 | 6±1 | 1.5±0.1 |
Total disagreement with Cooperativity of WT reported by Holroyde [1] and Smith [2] and very little Hill coefficient (nH) change with mutations. This is expected as only the subfragment S1 is used. It is S2 that binds the cMyBP-C. HCM, DCM mechanisms unclear.
DCM From several patients no protein mutation known
In samples from patients Wolff, et al. [50] report that the calcium concentration producing half-maximal tension ([Ca2+]50) was less in cardiomyopathic preparations than in donor preparations demonstrating an increase in myofibrillar calcium sensitivity of isometric tension which is lost on pKA phosphorylation. These results are contradictory to all other reports and probably reflect/suggest that the increased calcium sensitivity in this DCM report may be due at least in part to an in vivo reduction of the beta-adrenergically mediated phosphorylation of myofibrillar regulatory proteins such as cTnI and/or cMyBPC or their degradation.
The effects of HCM mutants of cTnI, in particular the large shifts in pCa50 for activation of the RCM mutants along with the two earlier indications of a concerted effect of cMyBP-C in conjunction with cTnI lead to a definite observation. The conclusion is together in the normal heart the cMyBP-C in conjunction with cTnI have the biochemical function of ensuring myosin bound MgATP is not the functional substrate of the cross-bridge ATPase. Disruption of either cTnI or cMyBP-C is sufficient to break this and allow MgATP to be used as substrate of the cross-bridge ATPase. The result of this is clear with a reduction in the Ca2+ cooperativity of activation and HCM. When unimpaired the joint action ensures that the system is fully relaxed in diastole on completion of the cycle with binding of MgATP. The cMyBP-C knock out myofibril does not display the commonly accepted cTnC affinity for Ca2+ it shows activation at higher [Ca2+]. Clearly one of the functions of cMyBP-C is to increase the calcium affinity of cTnC to that we are familiar with. The interaction with actin-myosin gives a change in Ca2+ sensitivity when an N-terminal fragment of cMyBP-C is added to the knock out myofibril. The phosphorylation state of the cTnI or others may have a bearing on this as well as the degree of pCa50 change with the mutants. The subtle interaction between cTnI and cMyBP-C is in need of much careful study. The overlap of the actin and myosin binding regions of cMyBP-C are certain candidates for the concerted action in substrate control. The possible binding of C1mC2 fragment of cMyBP-C to the actin along with the myosin is not clear in the literature reports. The effect of binding assorted segments of cMyBP-C in its absence may illuminate this.
The mechanisms of the effects, not arising from the cMyBP-C/cTnI system, of a range of mutations giving HCM and DCM are not clear. However when studying the response to calcium with an array of mutant sarcomeric proteins one may rationalise the effect of a mutation from the accepted function and interactions of the particular protein. Generally stronger interaction, more stress in filaments, more LIM release giving HCM and the reverse for DCM. Examples include floppy binding of MLC to the myosin giving DCM with retained ATPase function. Tropomyosin (α-TM) is involved in the interaction of the MLC with the actin filament, the crossbridge. All the HCM mutants of α-TM show a hindered dissociation of the crossbridge, slowed relaxation, resulting in transfer of chronic tension to the titin. Other α-TM mutants show reduced tension, at both or either [Ca2+]D and [Ca2+]sys resulting in DCM. TnT is directly involved with the stress transfer to the thin filament titin and hence mutations result in either HCM or DCM. Only one actin mutant is quoted giving weaker thin filament stress. Clues to the effect of mutations may be found in structural studies such as given by Lu, Wu and Morimoto, [51] and a review by de A. Marques M and de Oliveira [52].
I stress that all my conclusions regarding Ca2+ and Mg2+ in the myofibril are based entirely on the well respected measurement made on the intact system by Holroyde, Robertson, Johnson, Solaro and Potter, Morimoto and Ohtsuki [3], Donaldson, best and Kerrick [5], and myself et al. [2,4] and others in these references. To emphasise the need to only consider the intact system with all the many interactions of its constituents, I quote a more recent source Harris, Rostkova, Gautel and Moss14 who clearly demonstrate that the binding of cMyBP-C to the system has a major effect on the Ca2+ affinity of the cTnC, This is especially shown by the effect of adding the actin-myosin binding fragment of cMyBP-C (C1mC2) to the cMyBP-C knockout myofibrils.
A paper has just appeared that confuses the issue, but it can be ignored as all the measurements were performed on the Naked protein cTnC and a fragment of it, Rayani K, Seffernick JT, Li YA, Davis JP, Spuches AM, Van Petegem F, Solaro RJ, Lindert S, and Tibbits GF. Binding of Calcium and Magnesium to Cardiac Troponin C. doi: https://doi.org/10.1101/2020.06.14.150854. This article is a preprint and has not been certified by peer review.
There are no ethical considerations in this presentation and all quoted data is in the public domain and authors cited in each case.
- Holroyde MJ, Robertson SP, Johnson JD, Solaro RJ, Potter JD. The calcium and magnesium binding sites on cardiac troponin and their role in the regulation of myofibrillar adenosine triphosphatase. J Biol Chem. 1980; 25; 255: 11688–11693. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/6449512
- Smith GA, Dixon HB, Kirschenlohr HL, Grace AA, Metcalfe JC, Vandenberg JI. Ca2+ buffering in the heart: Ca2+ binding to and activation of cardiac myofibrils. Biochem J. 2000; 346 (Pt 2): 393–402. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1220865/
- Morimoto S and Ohtsuki I. Ca2+ binding to cardiac troponin C in the myofilament lattice and its relation to the myofibrillar ATPase activity. Eur J Biochem. 1994; 226: 597-602. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8001574
- Smith GA, Vandenberg JI, Freestone NS and Dixon HBF,The effect of Mg2+ on cardiac muscle function: is CaATP the substrate for priming myofibril cross-bridge formation and Ca2+ re-uptake by the sarcoplasmic reticulum? B J. 2001; 354: 539-551. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1221685/
- Donaldson SK, Best PM, Kerrick GL. Characterization of the effects of Mg2+ on Ca2+- and Sr2+-activated tension generation of skinned rat cardiac fibers. J Gen Physiol. 1978; 71: 645–655. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/97362
- Hofmann PA, Hartzell HC, Moss RL. Alterations in Ca2+ sensitive tension due to partial extraction of C-protein from rat skinned cardiac myocytes and rabbit skeletal muscle fibers. J Gen Physiol. 1991; 97: 1141–1163. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/1678777
- Kampourakis T, Yan Z, Gautel M, Sun YB and Irving M. Myosin binding protein-C activates thin filaments and inhibits thick filaments in heart muscle cells PNAS. 2014; 111: 18763-18768. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25512492
- Smith GA. Calcium, actomyosin kinetics, myosin binding protein-c and hypertrophic cardiomyopathy. J Integr Cardiol. 2019; 5: 1-2.
- Teekakirikul P, Zhu W, Huang HC, Fung E. Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules. 2019; 16 9: 878. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31888115
- van Dijk SJ, Dooijes D, dos Remedios C, Michels M , Lamers JMJ, et al. Cardiac Myosin-Binding Protein C Mutations and Hypertrophic Cardiomyopathy. Haploinsufficiency, Deranged Phosphorylation, and Cardiomyocyte Dysfunction. Circulation. 2009; 119: 1473–1483. PubMed:https://pubmed.ncbi.nlm.nih.gov/19273718/
- Doh CY, Li J, Mamidi R, Stelzer JE. The HCM-causing Y235S cMyBPC mutation accelerates contractile function by altering C1 domain structure. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2019; 1865: 661-677. PubMed:
- Parbhudayal RY, Garra AR, Götte MJW, Michels M, Pei J, et al. Variable Cardiac Myosin Binding protein-C Expression in the Myofilaments Due to MYBPC3 Mutations in Hypertrophic Cardiomyopathy. J Mol Cell Cardiol. 2018; 123: 59-63. PubMed: https://pubmed.ncbi.nlm.nih.gov/30170119
- Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, et al. Hypertrophic Cardiomyopathy in Cardiac Myosin Binding Protein-C Knockout Mice. Circ Res. 2002; 90: 594-601. PubMed: https://pubmed.ncbi.nlm.nih.gov/11909824
- Harris SP, Rostkova E, Gautel M, Moss RL. Binding of myosin binding protein-C to myosin subfragment S2 affects contractility independent of a tether mechanism. Circ Res.2004; 95. 930–936.
- Razumova MV, Bezold KL, Tu AY, Regnier M, Harris SP. Contribution of the Myosin Binding Protein C Motif to Functional Effects in Permeabilized Rat Trabeculae. JGen Physiol. 2008; 132: 575–585. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18955596
- Carrier L, Mearini G, Stathopoulou K, Cuello F. Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene. 2015; 573: 188-197. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6660134/
- Harris SP, Belknap B, Van Sciver RE, White HD, Galkin VE. C0 and C1 N-terminal Ig domains of myosin binding protein C exert different effects on thin filament activation. Proc Natl Acad Sci U S A. 2016; 113: 1558-1563.
- Mettikolla P, Calander N, Luchowski R, Gryczynski I, Gryczynski Z, et al. Cross-bridge Kinetics in Myofibrils Containing Familial Hypertrophic Cardiomyopathy R58Q Mutation in the Regulatory Light Chain of Myosin. J Theor Biol. 2011; 284: 71-81. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3152379/
- Lang R, Gomes AV, Zhao J, Housmans PR, Miller T, et al. Functional Analysis of a Troponin I (R145G) Mutation Associated with Familial Hypertrophic Cardiomyopathy. J Biol Chem. 2002; 277: 11670-11678. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11801593
- Davis J, Wen H, Edwards T, Joseph M, Metzger JM. Thin Filament Disinhibition by Restrictive Cardiomyopathy Mutant R193H Troponin I Induces Ca2+-Independent Mechanical Tone and Acute Myocyte Remodeling. Circulation Res. 2007; 100: 1494–1502. PubMed: https://pubmed.ncbi.nlm.nih.gov/17463320/
- Davis J, Wen H, Edwards T, Joseph M, Metzger JM. Allele and Species Dependent Contractile Defects by Restrictive and Hypertrophic Cardiomyopathy-Linked Troponin I Mutants. J Mol Cell Cardiol. 2008; 44: 891-904. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18423659
- Du J, Liu J, Feng HZ, Hossain MM, Gobara N, et al. Impaired relaxation is the main manifestation in transgenic mice expressing a restrictive cardiomyopathy mutation, R193H, in cardiac TnI Am J Physiol Heart Circ Physiol. 2008; 294: H2604–H2613. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2769498/
- Zhang J, Kumar A, Stalker HJ, Virdi G, Ferrans VJ, et al. Clinical and molecular studies of a large family with desmin-associated restrictive cardiomyopathy. Clin Genet. 200; 59: 248-256.
- Sheng JJ, Feng HZ, Pinto JR, Wei H, Jin JP. Increases of desmin and α-actinin in mouse cardiac myofibrils as a response to diastolic dysfunction. J Mol Cell Cardiol. 2016; 99: 218–229. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26529187
- Parvatiyar MS, Pinto JR, Dweck D, Potter JD. Cardiac Troponin Mutations and Restrictive Cardiomyopathy. J Biomed Biotech. 2010; 350706. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2896668/
- Jean-Charles PY, Li YJ, Nan CL, Huang XP. Monitoring Editor: Xiao Y-F. Insights into restrictive cardiomyopathy from clinical and animal studies. J Geriatr Cardiol. 2011; 8: 168–183. PubMed: https://pubmed.ncbi.nlm.nih.gov/22783303/
- Wen Y, Xu Y, Wang Y, Pinto JR, Potter JD, Kerrick WGL. Functional Effects of a Restrictive Cardiomyopathy linked Cardiac Troponin I mutation (R145W) in Transgenic Mice. J Mol Biol. 2009; 392: 1158–1167. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2774805/
- Gomes AV, Liang J, Potter JD. Mutations in Human Cardiac Troponin I That Are Associated with Restrictive Cardiomyopathy Affect Basal ATPase Activity and the Calcium Sensitivity of Force Development. J Biol Chem. 2005; 280: 30909-30915. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15961398
- Dyer EC, Jacques AM, Hoskins AC, Ward DG, Gallon CE, et al. Functional Analysis of a Unique Troponin C Mutation, GLY159ASP, that Causes Familial Dilated Cardiomyopathy, Studied in Explanted Heart Muscle. Circulation: Heart Failure. 2009; 2: 456–464.
- Swindle N, Albury ANJ, Baroud B, Burney M, Svetlana B, PubMed: Molecular and Functional Consequences of Mutations in the Central Helix of Cardiac Troponin C. Arch Biochem Biophys. 2014; 548: 46–53. PubMed: https://pubmed.ncbi.nlm.nih.gov/24650606/
- Keller DI, Coirault C, Rau T, Cheav T, Weyand M, et al. Human homozygous R403W mutant cardiac myosin presents disproportionate enhancement of mechanical and enzymatic properties. J Mol Cell Cardiol. 2004; 36: 355-62. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2440471/
- Kraft T, Witjas-Paalberends ER, Boontje NM, Tripathi S, Brandis A, et al Familial hypertrophic cardiomyopathy: Functional effects of myosin mutation R723G in cardiomyocytes. J Mol Cell Cardiol. 2013; 57: 13-22. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23318932
- Spudich JA, Aksel T, Bartholomew SR, Nag S, Kawana M, et al. Effects of hypertrophic and dilated cardiomyopathy mutations on power output by human β-cardiac myosin. J Exper Biol. 2016; 219: 161-167.
- Nag S, Sommese RF, Ujfalusi Z, Combs A, Langer S, et al. Parameters of human b-cardiac myosin with the hypertrophic cardiomyopathy mutation R403Q show loss of motor function. Sci Adv. 2015; 1: e1500511.
- Sarkar SS, Trivedi DV, Morck MM, Adhikari AS, Pasha SN, et al. The hypertrophic cardiomyopathy mutations R403Q and R663H increase the number of myosin heads available to interact with actin Sci Adv. 2020; 6: eaax0069. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/32284968
- Huang W, Liang J, Yuan CC, Kazmierczak K, Zhou Z, et al. Novel Familial Dilated Cardiomyopathy Mutation in MYL2 Affects the Structure and Function of Myosin Regulatory Light Chain. FEBS J. 2015; 282: 2379–2393. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4472530/
- Michele DE, Gomez CA, Hong KE, Westfall MV, Metzger JM. Cardiac Dysfunction in Hypertrophic Cardiomyopathy Mutant Tropomyosin Mice Is Transgene-Dependent, Hypertrophy-Independent, and Improved by β-Blockade. Circulation Res. 2002; 91: 255–262.
- Michele DE, Albayya FP, Metzger JM. Direct, Convergent Hypersensitivity of Calcium-Activated Force Generation Produced by Hypertrophic Cardiomyopathy Mutant Alpha-Tropomyosins in Adult Cardiac Myocytes. Nat Med. 1999; 5: 1413-1417. PubMed: https://pubmed.ncbi.nlm.nih.gov/10581085/
- Muthuchamy M, Pieples K, Rethinasamy P, Hoit B, Grupp IL, et al. Mouse Model of a Familial Hypertrophic Cardiomyopathy Mutation in α-Tropomyosin Manifests Cardiac Dysfunction. Circ. Res 1999; 85: 47–56. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10400910
- Chang AN, Harada K, Ackerman MJ, Potter JD. Functional consequences of hypertrophic and dilated cardiomyopathy-causing mutations in alpha-tropomyosin. J Biol Chem. 2005; 280: 34343-34349. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16043485
- Gupte TM, Haque F, Gangadharan B, Sunitha MS, Mukherjee S, et al. Mechanistic Heterogeneity in Contractile Properties of α-Tropomyosin (TPM1) Mutants Associated with Inherited Cardiomyopathies. J Biol Chem. 2015; 290: 7003-7015. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4358124/
- Rajan S, Ahmed RPH, Jagatheesan G, Petrashevskaya N, Boivin GP, et al. Dilated Cardiomyopathy Mutant Tropomyosin Mice Develop Cardiac Dysfunction With Significantly Decreased Fractional Shortening and Myofilament Calcium Sensitivity. Circulation Res. 2007; 101: 205–214. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17556658
- Song W, Dyer E, Stuckey D, Leung MC, Memo M, et al. Investigation of a transgenic mouse model of familial dilated cardiomyopathy. J. Mol Cell Cardiol. 2010; 49: 380–389. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20600154
- Hernandez OM, Szczesna-Cordary D, Knollmann BC, Miller T, Bell M, et al. F110I and R278C Troponin T Mutations That Cause Familial Hypertrophic Cardiomyopathy Affect Muscle Contraction in Transgenic Mice and Reconstituted Human Cardiac Fibers. J Biol Chem. 2005; 280: 37183-37194. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16115869
- Morimoto S, Lu QW, Harada K, Takahashi-Yanaga F, Minakami R, et al. Ca2+-desensitizing effect of a deletion mutation ΔK210 in cardiac troponin T that causes familial dilated cardiomyopathy PNAS. 2002; 99: 913-918.
- Sfichi-Duke L, Garcia-Cazarin ML, Sumandea CA, Sievert GA, Balke CW, et al. Cardiomyopathy-causing Deletion K210 in Cardiac Troponin T Alters Phosphorylation Propensity of Sarcomeric Proteins. J Mol Cell Cardiol. 2010; 48: 934-942.
- Venkatraman G, Gomes AV, Kerrick WGL, Potter JD. Characterization of Troponin T Dilated Cardiomyopathy Mutationsin the Fetal Troponin Isoform. J Biol Chem. 2005; 280: 17584–17592. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15623536
- Jones KmD, Koh Y, Weller RS, Turna RS, Ahmad F, et al. Pathogenic Troponin T Mutants With Opposing Effects on Myofilament Ca 2+ Sensitivity Attenuate Cardiomyopathy Phenotypes in Mice. Arch Biochem Biophys. 2019; 661: 125-131. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30445044
- Sommese RF, Nag S, Sutton S, Miller SM, Spudich JA, et al. Effects of Troponin T Cardiomyopathy Mutations on the Calcium Sensitivity of the Regulated Thin Filament and the Actomyosin Cross-Bridge Kinetics of Human β-Cardiac Myosin. PLOS ONE. 2013; 8: e83403. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3867432/
- Wolff MR, Buck SH, Stoker SW, Greaser ML, Mentzeri RM. Myofibrillar Calcium Sensitivity of Isometric Tension Is Increased in Human Dilated cardiomyopathies Role of Altered b-adrenergically Mediated Protein Phosphorylation. J Clin Invest. 1996; 98: 167–176. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8690789
- Lu QW, Wu XY, Morimoto S. Inherited cardiomyopathies caused by troponin mutations. J Geriatr Cardiol. 2013; 10: 91–101. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23610579
- de A Marques M, de Oliveira GAP. Cardiac Troponin and Tropomyosin: Structural and Cellular Perspectives to Unveil the Hypertrophic Cardiomyopathy Phenotype. Front Physiol. 2016; 7: 429. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27721798