Mini Review
Thrombolysis, the only Optimally Rapid Reperfusion Treatment

Victor Gurewich*
Vascular Research Laboratory, Mount Auburn Hospital, Cambridge, MA 02138, Harvard Medical School, USA
*Address for Correspondence: Victor Gurewich, Vascular Research Laboratory, Mount Auburn Hospital, Cambridge, MA 02138, Harvard Medical School, USA, Email: vgurewich@tsillc.net
Dates: Submitted: 29 May 2017; Approved: 22 June 2017; Published: 23 June 2017
How to cite this article: Gurewich V. Thrombolysis, the only Optimally Rapid Reperfusion Treatment. J Cardiol Cardiovasc Med. 2017; 2: 029-034.
DOI: 10.29328/journal.jccm.1001010
Copyright License: © 2017 Gurewich V. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
Thrombolysis with tissue plasminogen activator (tPA) has been plagued by inadequate efficacy and a high risk of intracranial hemorrhage (ICH), which led to its replacement by procedures like percutaneous coronary intervention (PCI) whenever possible. Since this requires hospitalization, it is time-consuming, and compromising salvage of brain tissue and myocardium. Thrombolysis is the only first-line treatment that can provide sufficiently timely treatment for optimal recovery of organ function. However, for this potential to be realized, its efficacy and safety must be significantly improved over the current method. By adopting the sequential, synergistic fibrinolytic paradigm of the endogenous system, already verified by a clinical trial, this becomes possible. The endogenous system’s function is evidenced by the fibrinolytic product D-dimer that is invariably present in blood, and which increases >20-fold in the presence of thromboembolism. This system uses tPA to initiate lysis, which is then completed by the other fibrin-specific activator prourokinase (proUK). Since tPA and proUK in combination are synergistic in fibrinolysis, it helps explain their efficacy at their low endogenous concentrations.
INTRODUCTION
Thrombolysis has been dominated by the use of tPA alone predicated on the conviction this activator was responsible for fibrinolysis. This assumption was maintained despite the first comparative trials against the original clinical plasminogen activator, streptokinase (SK), contradicting all expectations.
In this pivotal first study, three mega trials were required involving an unprecedented total of 95,740 patients with acute myocardial infarction (AMI) to achieve a statistically significant difference between fibrin-specific tPA and non-specific SK. In the first two trials, the mortalities in the two treatment groups was identical [1, 2]. In the last trial, in only one of four groups was there a statistically significant lower 30 day mortality (6.9 v.s 7.8%) with tPA [3]. This small difference did not stand up to a Bayesian statistical analysis, which concluded that a real difference between tPA and SK had not been established [4]. Nevertheless, tPA was approved and soon became the activator of choice even though it caused significantly more intracranial hemorrhage (ICH) than SK [2,3], a finding that also contradicted expectations.
No explanation for these unexpected results was offered, and the investigators concluded that “mortality differences…from different fibrinolytic regimens are unlikely to be large” [2]. As a result, interest in thrombolysis within the scientific community and the pharmaceutical industry faded after these trials, and thrombolytic development came to a virtual standstill.
After years of more disappointing clinical benefits from tPA, it was replaced by percutaneous coronary intervention (PCI) whenever possible, and primary PCI has become the treatment of choice for AMI, and endovascular procedures are also being used in ischemic stroke whenever possible. Endovascular procedures have proven to be more beneficial than tPA in comparative trials, but since they require hospitalization, they inevitably delay the reperfusion of ischemic tissue. For other indications like pulmonary thromboembolism, tPA is used only in patients with hemodynamic instability due to tPA’s bleeding hazard.
The mega trial results can be explained by tPA’s restricted mechanism of plasminogen activation
The high fibrin affinity of tPA, the property responsible for its fibrin specificity and endowed it with high clinical expectations, also restricts its fibrin-dependent plasminogen activation. When tPA binds to fibrin, it is at a single binding site on the D-domain of intact fibrin adjacent to a plasminogen binding site on Lysine Aα-157 [5]. The resulting fibrin-plasminogen-tPA ternary complex promotes tPA’s activation of this plasminogen about 1,000-fold [6], a highly efficient mechanism that is responsible for initiating fibrin degradation [5-7]. However, this efficient plasminogen activation is limited to this one site.
Fibrinolysis requires the activation of the remaining fibrin-bound plasminogens which are two in number [8]. Against these plasminogens that are not in the ternary complex, tPA is a weak plasminogen activator, being about 1,000-fold less active [6], explaining why such high tPA doses are required for efficacy. By contrast, SK is an unrestricted activator and is equally active against all plasminogens. Their net clinical effects turned out to be comparable.
The high doses of tPA required for efficacy when it is used alone resulted in more ICH complications than with non-fibrin-specific SK [1-3]. This bleeding side effect is due to tPA’s lysis of hemostatic fibrin [9]. This is because hemostatic fibrin contains the tPA and plasminogen binding sites on the D-domain of intact fibrin where the ternary complex is formed [6]. Such hemostatic fibrin is present at occult sites that are believed to represent repair sites from wear-and-tear vascular injuries.
Therefore, both tPA’s limited beneficial effects and bleeding risk seen in the mega trials can be explained by its mechanism of fibrin-specific plasminogen activation. When thrombolysis was eventually abandoned due to the disappointing results of tPA given alone, this was somewhat akin to the proverbial throwing out the baby, thrombolysis, with the bathwater, tPA. This is because tPA alone is not the same as thrombolysis, since tPA is only the initiator of thrombolysis, analogous to the starter mechanism of an automobile.
Due to the technically demanding nature of PCI, it can only be a limited solution for a ubiquitous conditions like AMI and stroke. In addition, it tends to delay reperfusion beyond the time when salvage of most myocardial or brain tissue is possible. The greatest reduction in AMI mortality and post AMI morbidity is achieved when coronary reperfusion takes place within 1-2 hours of symptom onset [10,11], and this event to reperfusion time can only be achieved by thrombolysis for the vast majority of patients.
How thrombolysis can be improved?
The finding that tPA alone was insufficient for effective thrombolysis implicates the other biological plasminogen activator, proUK. Both are involved in the endogenous fibrinolytic system. Since this system functions with a tPA concentration three orders of magnitude less than that needed for therapy, half of which circulates as an inactive inhibitor complex [13], it is evident that tPA cannot alone be responsible for endogenous fibrinolysis.
Evidence of the efficacy of this system comes from the presence in blood of the fibrinolytic product D-dimer. Its invariable presence in the plasma of even healthy individuals (normal range: 112-250 ng/mL) indicates that fibrinolysis is ongoing. In the presence of venous thromboembolism, the D-dimer level goes up to ≥5,000 ng/mL, showing that the system has a significant reserve capacity. Only in a patient with an unusually potent auto-antibody to thrombin has the normal D-dimer concentration been reported to be nearer zero (12-32 ng/mL) [14], a finding that also confirms that fresh intravascular fibrin, whose formation was suppressed by the antibody, is the D-dimer’s source.
Fibrinolysis induced by such low tPA concentrations can be explained by the complementary modes of action of tPA and proUK [15] and their fibrinolytic effect which when combined is synergistic [16]. Unlike tPA that is stored in the vessel wall, from where it initiates fibrinolysis, proUK is a is a normal constituent of blood, from where it continues fibrinolysis. Since most proUK is bound to platelets [17,18] and monocytes [19], rather than being free in plasma, it has often been overlooked. In addition, proUK is inactivated by thrombin so that when it is experimentally included in clot formation, it can be destroyed and escape detection. These are some of the factors that have contributed to the commonly held but erroneous belief that tPA alone was responsible for intravascular fibrinolysis.
Furthermore, when the proUK cell receptor (UPAR) was identified, which enables proUK to induce pericellular plasminogen activation for cell migration, it was concluded that proUK is exclusively an extravascular rather plasminogen activator [12]. Although this concept was contradicted by other evidence, it has persisted to the present [20].
Evidence to the contrary comes not only from the complementary modes of action of the two activators but also includes gene knockout studies. These showed that the deletion of tPA had surprisingly little effect on the lysis of a venous thrombus in mice. By contrast, a proUK knockout suppressed clot lysis markedly [21]. The more potent effect of proUK is consistent with its two-fold lytic effect since proUK is activated to its enzymatic form, urokinase (UK), during fibrinolysis which activates an additional plasminogen. The conversion of single-chain tPA to its two-chain form does not change its activity or fibrinolytic effect [22]. Due to this double effect of proUK/UK, the clot lysis rate by proUK at a fibrin-specific dose was consistently about two-fold greater than that by tPA [23].
Since proUK has no fibrin affinity and is dependent on plasminogen bound to partially the degraded fibrin on the E‑domain [7], proUK spares hemostatic fibrin which is intact fibrin. This property along with it being a proenzyme, makes proUK well suited for therapeutic administration as an infusion. By contrast, since tPA is an active enzyme with an exceptionally high fibrin affinity [24], it is much better suited for a bolus administration, as it is used in endogenous fibrinolysis. Since tPA’s fibrinolytic function is the initiation of fibrinolysis [5-7], this may also be sufficient as long as it is not given alone (Figure 1).
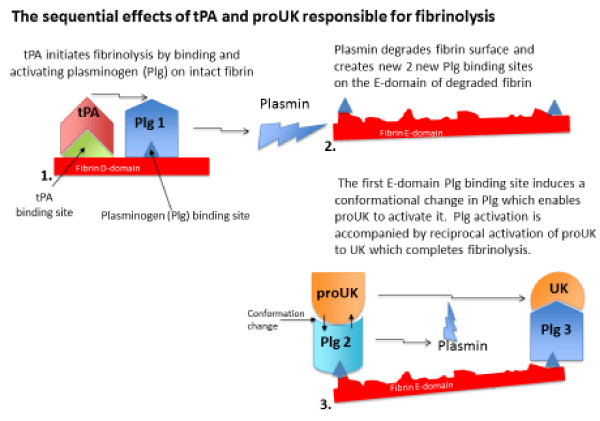
Figure 1: Fibrinolysis is initiated when tPA released from the vessel wall binds to its binding site on the D-domain of intact fibrin and activates plasminogen bound to an adjacent site. This ternary complex with fibrin promotes tPA plasminogen activation by about 1,000-fold. Fibrin degradation creates two new plasminogen binding sites on the fibrin E-domain. The first of these is a triple carboxyl lysine binding site which induces a unique conformational change in plasminogen that enables the intrinsic activity of proUK to activate it. This is accompanied by reciprocal activation of proUK to UK which activates the remaining plasminogen completing fibrinolysis.
In view of the complementary properties of tPA and proUK and their complementary functions in fibrinolysis [15], a logical solution for improving thrombolysis is by using a combination of both plasminogen activators, as in physiological fibrinolysis.
Sequential plasminogen activation by tPA and proUK in endogenous fibrinolysis
When an intravascular thrombus forms, tPA in the vessel wall at that site is released, and due to its high fibrin affinity, it binds to the thrombus and initiates fibrin degradation. This creates two new plasminogen binding sites on the fibrin surface of the clot [24]. The first of these is a triple C-terminal lysine binding site on the fibrin E-domain, and when plasminogen binds to this site it undergoes a unique conformational change which enables its activation by the intrinsic activity of proUK [25]. Activation of this plasminogen is accompanied by reciprocal activation of proUK to UK by plasmin [26], and UK then activates the remaining fibrin-bound plasminogen to complete fibrinolysis.
Therefore, fibrinolysis takes place sequentially by the activators, since tPA is required for the initiation of fibrinolysis and proUK/UK for its continuation and completion (Figure 1). Such a regimen was put to a clinical test.
Thrombolysis by the endogenous paradigm of fibrinolysis
In the PATENT trial, 101 patients with AMI were treated with an initial mini-bolus of tPA followed by an i.v. infusion of proUK. In the first 10 patients, a 10 mg bolus was given which was found to be excessive, so the remaining 91 patients received only a 5 mg tPA bolus (5% of its monotherapy dose). This was followed by an i.v. infusion of proUK, 40 mg/h for 90 minutes (50% of its monotherapy infusion rate) [27].
This regimen induced complete coronary artery patency (TIMI-3) at 24h in 82% of the patients, which compares with a 45% TIMI-3 rate at 24h in the best of the tPA trials, the GUSTO trial [28]. The high patency rate in PATENT was associated with an exceptionally low mortality of 1%, which compares with a 6.3% mortality in GUSTO [3].
Unfortunately, no subsequent clinical trials of this regimen were possible since proUK development was abandoned a few years after this trial. The reason for this was that in trials of proUK given alone, in which a two-fold higher infusion rate was necessary, proUK activated plasma plasminogen to plasmin, and plasmin activated proUK to UK. Since UK is a non-fibrin-specific activator, it caused bleeding side effects. As a result proUK was denied approval by the European regulatory agency, the EMEA.
In conclusion, fibrinolysis occurs as a result of the sequential, complementary, and synergistic effects of both natural plasminogen activators in combination. Effective and safe thrombolysis cannot be achieved by either activator alone. By this natural combination of both activators, thrombolysis can provide more rapid reperfusion of occluded arteries than is possible by current methods, and thereby achieve a better recovery of organ function and a lower mortality.
REFERENCES
- GISSI-2: a factorial randomised trial of alteplase versus streptokinase and heparin versus no heparin among 12,490 patients with acute myocardial infarction. Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto Miocardico. Lancet. 1990; 336: 65-71. Ref.: https://goo.gl/5BcND7
- ISIS-3: a randomised comparison of streptokinase vs tissue plasminogen activator vs anistreplase and of aspirin plus heparin vs aspirin alone among 41,299 cases of suspected acute myocardial infarction. ISIS-3 (Third International Study of Infarct Survival) Collaborative Group. Lancet 1992; 339: 753-770. Ref.: https://goo.gl/cNYjDk
- GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Eng J Med 1993; 329: 673-682. Ref.: https://goo.gl/tDc4Tb
- Brophy JM, Joseph L. Placing trials in context using Bayesian analysis. GUSTO revisited by Reverend Bayes. JAMA 1995; 273: 871-875. Ref.: https://goo.gl/j3PpnF
- Voskuilen M, Vermond A, Veeneman GH, van Boom JH, Klasen EA. Fibrinogen lysine residue Aα157 plays a crucial role in the fibrin-induced acceleration of plasminogen activation, catalyzed by tissue-type plasminogen activator. J Biol Chem. 1987; 262; 5944-5946. Ref.: https://goo.gl/HNvZ3H
- Hoylaerts M, Rijken DC, Lijnen HR, Collen D. Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role of fibrin. J Biol Chem. 1982; 257: 2912-2919. Ref.: https://goo.gl/jFRdfA
- Liu JN, Gurewich V. A comparative study of the promotion of tissue plasminogen activator and pro-urokinase-induced plasminogen activation by fragments D and E-2 of fibrin. J Clin Invest. 1991; 88: 2012-2017. Ref.: https://goo.gl/NohYzy
- Suenson E, Lützen O, Thorsen S. Initial plasmin-degradation of fibrin as the basis of a positive feed-back mechanism in fibrinolysis. Eur J Biochem. 1984; 140: 513-5229. Ref.: https://goo.gl/nucZSA
- Marder VJ, Sherry S. Thrombolytic therapy: current status. N Engl J Med 1988; 318: 1512-1520. Ref.: https://goo.gl/pComu6
- Boersma E, Maas ACP, Deckers JW, Simoons ML. Early thrombolytic treatment in acute myocardial infarction: reappraisal of the golden hour. Lancet. 1996; 348: 771-775. Ref.: https://goo.gl/LK4akV
- Schofield PM. Acute myocardial infarction: the case for pre-hospital thrombolysis with or without percutaneous coronary intervention. Heart. 2005; 91: 7-11. Ref.: https://goo.gl/YTrUh8
- Collen D. The plasminogen (fibrinolytic) system. Thromb Haemost. 1999; 82: 259-270. Ref.: https://goo.gl/y7fZSc
- Chandler WL, Jascur ML, Henderson PJ. Measurement of different forms of tissue plasminogen activator in plasma. Clin Chem. 2000; 46: 38-46. Ref.: https://goo.gl/J2j26v
- Baglin TP, Landown R, Frasson R, Huntington JA. Discovery and characterization of an antibody directed against exosite 1 of thrombin. J Thromb Haemos. 2016; 4: 137-142. Ref.: https://goo.gl/EJJEmS
- Pannell R, Black J, Gurewich V. The complementary modes of action of tissue plasminogen activator (t-PA) and pro-urokinase (pro-UK) by which their synergistic effect on clot lysis may be explained. J Clin Invest. 1988; 81: 853-859. Ref.: https://goo.gl/c3zdcV
- Gurewich V, Pannell R. Synergism of tissue-type plasminogen activator (t-PA) and single-chain urokinase-type plasminogen activator (scu-PA) on clot lysis in vitro and a mechanism for this effect. Thromb Haemost. 1987; 57: 372-378. Ref.: https://goo.gl/GgWwzV
- Vaughan DE, Vautloutle E, Collen D. Urokinase binds to platelets through a specific saturable, low affinity mechanism. Fibrinolysis. 1990; 4: 141. Ref.: https://goo.gl/hbESgz
- Gurewich V, Johnstone MT, Pannell R. The selective uptake of high molecular weight urokinase-type plasminogen activator by human platelets. Fibrinolysis. 1995; 9: 188-195. Ref.: https://goo.gl/KHGDQQ
- Grau E, Moroz LA. Fibrinolytic activity of normal human blood monocytes. Thromb Res. 1989; 53: 145-162. Ref.: https://goo.gl/RqjzL9
- Longstaff C, Kolev K. Basic mechanisms and regulation of fibrinolysis. J Thromb Haemostas. 2015; 13: 98-105. Ref.: https://goo.gl/UckqPw
- Singh I, Burnand KG, Collins M, Luttun A, Collen D, et al. Failure of thrombus to resolve in urokinase-type plasminogen activator gene-knockout mice: rescue by normal bone marrow-derived cells. Circulation. 2003; 107: 869-875. Ref.: https://goo.gl/Wsb6M2
- Rijken DC, Hoylaerts M, Collen D. Fibrinolytic properties of one-chain and two-chain human extrinsic (tissue-type) plasminogen activator. J Biol Chem. 1982; 257: 2920-2925. Ref.: https://goo.gl/yhRLr3
- Pannell R, Gurewich V. A comparison of the rates of clot lysis in a plasma milieu induced by tissue plasminogen activator (t-PA) and rec-prourokinase: evidence that t-PA has a more restricted mode of action. Fibrinolysis. 1992; 6: 1-5. Ref.: https://goo.gl/7WYcct
- Harpel PC, Chang TS, Verderber E. Tissue plasminogen activator and urokinase mediate the binding of Glu-plasminogen to plasma fibrin I. Evidence for new binding sites in plasmin-degraded fibrin I. J Biol Chem. 1985; 260: 4432-4430. Ref.: https://goo.gl/cGwU86
- Liu J, Gurewich V. Fragment E-2 from fibrin substantially enhances pro-urokinase-induced glu-plasminogen activation. A kinetic study using a plasmin-resistant mutant pro-urokinase Ala-158-rpro-UK. Biochemistry. 1992; 31: 6311-6317. Ref.: https://goo.gl/5cRPnh
- Petersen LC. Kinetics of reciprocal pro-urokinase/plasminogen activation. Stimulation by a template formed by the urokinase receptor bound to poly (D-lysine). Eur J Biochem. 1997; 245: 316-323. Ref.: https://goo.gl/nkfoqD
- Zarich SW, Kowalchuk GJ, Weaver WD, Loscalzo J, Sassower M, Manzo K, Byrnes C, Muller JE, Gurewich V for the PATENT Study Group. Sequential combination thrombolytic therapy for acute myocardial infarction: results of the pro-urokinase and t-PA enhancement of thrombolysis (PATENT) trial. J Am Coll Cardio. 1995; 26: 374-379. Ref.: https://goo.gl/qHExpB
- The GUSTO Angiographic Investigators. The effects of tissue plasminogen activator, streptokinase, or both on coronary-artery patency, ventricular function, and survival after acute myocardial infarction. N Engl J Med. 1993; 329: 1615-1622. Ref.: https://goo.gl/NkBbWC